
The 10 Phases Of An Effective CAPA
By Mark Durivage, Quality Systems Compliance LLC
Corrective and preventive action (CAPA) issues continue to be one of the top Form 483 observational findings by the FDA. Many times, CAPAs fail due to the structure and flow of the process and not necessarily the efforts of those managing the CAPAs. A poor paper-based CAPA process will not improve with an electronic-based CAPA system. Oftentimes, organizations do not fully document the CAPA phases and confuse verification of implementation and verification of implantation activities. This article will look at some of the regulations governing CAPAs and discuss the elements of 10 phases of a CAPA investigation that will help organizations ensure their CAPA processes are destined to succeed.
FDA. Many times, CAPAs fail due to the structure and flow of the process and not necessarily the efforts of those managing the CAPAs. A poor paper-based CAPA process will not improve with an electronic-based CAPA system. Oftentimes, organizations do not fully document the CAPA phases and confuse verification of implementation and verification of implantation activities. This article will look at some of the regulations governing CAPAs and discuss the elements of 10 phases of a CAPA investigation that will help organizations ensure their CAPA processes are destined to succeed.
CAPA Requirements
21 CFR 820 Quality System Regulation
Sec. 820.100 Corrective and preventive action
(a) Each manufacturer shall establish and maintain procedures for implementing corrective and preventive action. The procedures shall include requirements for:
(1) Analyzing processes, work operations, concessions, quality audit reports, quality records, service records, complaints, returned product, and other sources of quality data to identify existing and potential causes of nonconforming product, or other quality problems. Appropriate statistical methodology shall be employed where necessary to detect recurring quality problems;
(2) Investigating the cause of nonconformities relating to product, processes, and the quality system;
(3) Identifying the action(s) needed to correct and prevent recurrence of nonconforming product and other quality problems;
(4) Verifying or validating the corrective and preventive action to ensure that such action is effective and does not adversely affect the finished device;
(5) Implementing and recording changes in methods and procedures needed to correct and prevent identified quality problems;
(6) Ensuring that information related to quality problems or nonconforming product is disseminated to those directly responsible for assuring the quality of such product or the prevention of such problems; and
(7) Submitting relevant information on identified quality problems, as well as corrective and preventive actions, for management review.
(b) All activities required under this section, and their results, shall be documented.
ISO 13485:2016 Medical devices — Quality management systems — Requirements for regulatory purposes
8.5.2 Corrective action
The organization shall take action to eliminate the cause of nonconformities in order to prevent recurrence. Any necessary corrective actions shall be taken without undue delay. Corrective actions shall be proportionate to the effects of the nonconformities encountered.
The organization shall document a procedure to define requirements for:
a) reviewing nonconformities (including complaints);
b) determining the causes of nonconformities;
c) evaluating the need for action to ensure that nonconformities do not recur;
d) planning and documenting action needed and implementing such action, including, as appropriate, updating documentation;
e) verifying that the corrective action does not adversely affect the ability to meet applicable regulatory requirements or the safety and performance of the medical device;
f) reviewing the effectiveness of corrective action taken.
Records of the results of any investigation and of action taken shall be maintained.
8.5.3 Preventive action
The organization shall determine action to eliminate the causes of potential nonconformities in order to prevent their occurrence. Preventive actions shall be proportionate to the effects of the potential problems.
The organization shall document a procedure to describe requirements for:
a) determining potential nonconformities and their causes;
b) evaluating the need for action to prevent occurrence of nonconformities;
c) planning and documenting action needed and implementing such action, including, as appropriate, updating documentation;
d) verifying that the action does not adversely affect the ability to meet applicable regulatory requirements or the safety and performance of the medical device;
e) reviewing the effectiveness of the preventive action taken, as appropriate.
Records of the results of any investigations and of action taken shall be maintained.
The Joint IPEC - PQG Good Manufacturing Practices Guide for Pharmaceutical Excipients
8.5.2 Corrective Action
The excipient manufacturer should establish, document and maintain procedures for:
- determining the root causes of nonconformities,
- ensuring that corrective actions are implemented and effective,
- implementing and recording changes in procedures resulting from corrective action.
8.5.3 Preventive Action
The excipient manufacturer should establish, document and maintain procedures for:
- initiating preventive actions to deal with problems at a level corresponding to the risks,
- implementing and recording changes in procedures resulting from preventive action.
ICH Harmonised Tripartite Guideline Pharmaceutical Quality System Q10
3.2.2 Corrective Action and Preventive Action (CAPA) System
The pharmaceutical company should have a system for implementing corrective actions and preventive actions resulting from the investigation of complaints, product rejections, non-conformances, recalls, deviations, audits, regulatory inspections and findings, and trends from process performance and product quality monitoring. A structured approach to the investigation process should be used with the objective of determining the root cause. The level of effort, formality, and documentation of the investigation should be commensurate with the level of risk, in line with ICH Q9. CAPA methodology should result in product and process improvements and enhanced product and process understanding.
CAPA Phases
A good CAPA process consists of 10 distinct phases, as shown in the figure below.

1) Problem Identification and CAPA Initiation
The problem identification and CAPA initiation phase requires documenting the issue to begin the CAPA process. The description should be as complete as possible, including who, what, when, where, why, and how many.
2) Risk Analysis
A risk analysis should be performed based on patient/user/business/compliance risk. The results of the risk analysis should drive CAPA timelines. It should be obvious that low-risk issues do not have the same sense of urgency as high-risk issues.
3) Correction/Containment
Correction and containment should be completed as soon as possible to prevent further production and distribution. The organization should review related processes and products to see if there are wider issues. In the case of product-related issues, a field correction and/or recall may be required. Correction and containment activities are an effort to reduce the impact in the short term.
Correction activities can include sorting, reinspection, reworking, and reprocessing.
4) Investigation/Root Cause Analysis
Several of the most common tools/methods for performing investigations to determine the root cause of a problem include but are not limited to:
• Brainstorming
• The 5 whys
• Flowcharting
• Fishbone diagrams
• Affinity diagrams
• Is/Is Not
• TRIZ (Theory of Inventive Problem-Solving)
• Physics of Failure
5) Corrective/Preventive Action(s)
Corrective and preventive actions are considered long-term solutions to resolve or eliminate the cause of a nonconformity or eliminate the cause of a potential nonconformity. A corrective action is an action to eliminate the cause of a nonconformity and to prevent recurrence. A preventive action is an action to eliminate the cause of a potential nonconformity or other potential undesirable situation. A correction is an action to eliminate a detected nonconformity.
6) Implementation
The implementation phase is where corrective and preventive actions are initiated and applied to address the root cause or causes of a nonconformity. Examples are procedural updates, training, and process modifications.
7) Verification of Implementation
Verification is defined as confirmation, through the provision of objective evidence that specified requirements have been fulfilled. The verification of implementation phase documents that corrective and preventive actions were actually implemented.
The verification of implementation (VOI) phase is often confused with the verification of effectiveness (VOE) phase of a CAPA, but these are separate, distinct phases.
VOIs are used to verify that corrections, containment, corrective actions, and preventive actions were indeed implemented as promised and planned.
Examples of VOIs include:
• Procedures, work instructions, forms, and/or templates were updated.
• A change order was processed
• The individual(s) were trained or retrained
• The suspect parts were sorted
• The suspect parts were reworked
• The suspect parts were quarantined
8) Verification of Effectiveness Plan (VOEP)
The effectiveness plan phase is used to establish and define predetermined criteria to verify that corrective and/or preventive actions were indeed effective.
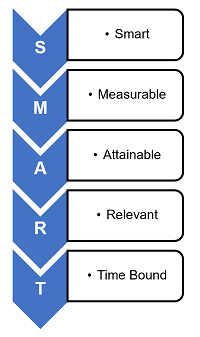
George T. Doran is credited with first using the term SMART:
- Specific — Is the VOEP unambiguous, clear, and focused?
- Measurable — Is quantifiable data being used to assess the VOEP?
- Achievable — Is the VOEP feasible or practical?
- Relevant — Is the VOEP appropriate for the level of risk?
- Time bound — Does the VOEP have a realistic deadline?
Peter Drucker's management by objectives concept utilized SMART goals. The SMART methodology is usually associated with personal goals related to annual bonus, but it can be readily be applied to CAPA VOEP activities.
Examples of a VOEP’s checks include:
- The problem has not recurred in the last three production lots
- The problem has not recurred in the last three months
- The first pass yield is >99 percent
- The scrap rate will decrease by 5 percent (say, from 15 percent to 10 percent)
9) Verification of Effectiveness (VOE)
The verification of effectiveness phase includes reviewing and documenting the predetermined criteria established in the verification of effectiveness plan. A successful verification of effectiveness should demonstrate the true root cause of the issue was properly identified and the corrective and/or preventive actions were effective.
Examples of a VOE’s checks and corresponding verifications include:
- The problem has not recurred in the last three production lots. Verification: Lots #123, #456, #789 were reviewed to demonstrate the CAPA was effective.
- The problem has not recurred in the last three months. Verification: The complaint log and non-conformance report (NCR) log were reviewed to demonstrate the CAPA was effective.
- The first pass yield is >99 percent. Verification: The first pass yield is 99.25, percent, which demonstrates the CAPA was effective.
- The scrap rate will decrease by 5 percent (from 15 percent to 10 percent). Verification: The scrap rate is now 9.5 percent, which demonstrates the CAPA was effective.
10) Closure
This is the last phase of the CAPA process. A CAPA should only be closed once the verification of effectiveness has been successfully completed. If a CAPA was found to be ineffective, I suggest opening a new CAPA and referencing the original CAPA.
Conclusion
Not only are CAPAs required by regulations, standards, and guidances, CAPAs, if performed correctly, can help an organization improve its competitive position by reducing waste and improving processes. Each phase of a CAPA should be an independent section on a form or requirement in an electronic workflow to ensure the process phases are well defined.
About The Author:
 Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is managing principal consultant at Quality Systems Compliance LLC, an ASQ Fellow, and an SRE Fellow. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. He holds several certifications, including CRE, CQE, CQA, CSQP, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. Durivage resides in Lambertville, Michigan. Please feel free to email him at mark.durivage@qscompliance.com or connect with him on LinkedIn.
Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is managing principal consultant at Quality Systems Compliance LLC, an ASQ Fellow, and an SRE Fellow. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. He holds several certifications, including CRE, CQE, CQA, CSQP, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. Durivage resides in Lambertville, Michigan. Please feel free to email him at mark.durivage@qscompliance.com or connect with him on LinkedIn.