
Novel Approaches For Obtaining High-productivity Clones
By Susan Sharfstein, professor of nanoscale science and engineering, University at Albany

As therapeutic proteins play an increasingly important role in healthcare, with an average of 14 new therapeutic proteins approved each year over the last seven years and 45 biosimilar approvals since 2015, cell line development continues to be a challenging, labor-intensive process. A large number of technologies have been introduced to improve clone screening and verification of monoclonality,1 reducing the manual labor of limited-dilution cloning and microscopic cell screening; however, these technologies have not improved the quality of the initial pool of cells to sort or reduced the time required for antibiotic or metabolic selection.
In this article, I will discuss novel approaches to developing highly productive clones, including a novel cell line selection approach based on post-transcriptional selection, the use of transposons to increase copy number for the gene of interest, and targeted integration to place the transgenes into “hot spot” loci in the chromosome to rapidly obtain high productivity clones. Future articles in this series will address gene editing technologies such as CRISPR and zinc finger nucleases for host cell engineering and strategies for production of difficult to express proteins.
Challenges And Opportunities In The Production Of Recombinant Therapeutic Proteins
Currently, the vast majority of therapeutic proteins are produced in Chinese hamster ovary cells, generally using one of two metabolic selection strategies: complementation of a deficiency of dihydrofolate reductase (DHFR) using methotrexate (MTX) for selection or complementation of a deficiency of glutamine synthetase (GS) using methionine sulfoximine (MSX). Antibiotic selection (e.g., neomycin, hygromycin, blasticidin) is sometimes used as an alternative or supplement (e.g., in two vector systems) to metabolic selection. In general, the transgenes are introduced by either chemical transfection or electroporation, using either circular or linearized plasmids. There is increasing interest in viral transduction; however, this approach brings with it increased risks of viral contamination of the product and limitations in the size of the DNA that can be incorporated, so it is not widely used. In the case of metabolic selection, transfection and selection are often followed by an increase in selection pressure by increasing the concentration of MTX or MSX to increase the gene copy number, sometimes after an initial round of clonal selection. Finally, regardless of transfection or selection method, cells are then clonally isolated and screened to identify high-productivity clones with good growth properties.
While the aforementioned techniques are quite effective at introducing heterologous DNA into cells, they suffer from a number of challenges. First, the rate of stable integration of the DNA into cells is low. Moreover, the DNA is randomly integrated into the chromosome during the transfection/selection process with no targeting to loci that are transcriptionally accessible or highly transcribed, creating substantial heterogeneity between transfected cells. This random integration can also lead to instability of both the gene and transcription as some sites are prone to recombination and/or to epigenetic silencing. Second, there is no control of gene copy number, and in particular, for two vector systems (heavy chain and light chain on different plasmids), the levels of heavy chain (HC) polypeptide and light chain (LC) polypeptide may become unbalanced. Finally, regardless of whether metabolic or antibiotic selection are used, in both cases, the selection is based upon the transcription, translation, folding, and activity of a protein entirely distinct from the gene of interest. Despite the development of vectors with the DHFR or GS gene under the control of weaker promoters, this approach leads to a waste of resources in producing these metabolic selection proteins, and often the ability to withstand higher concentrations of selection reagent (e.g., MTX, neo, etc.) does not lead to higher production of the transgene. Recently, a variety of new technologies have attempted to address some of the issues raised above.
Novel Cell Line Selection Based On Post-transcriptional Selection
In conjunction with industrial collaborators, my laboratory demonstrated the ability to select cells expressing the gene of interest using a silencing-RNA (siRNA) that is co-transcribed with the gene of interest (GOI). In this novel approach, a cassette containing the siRNA is located in the 5’ untranslated region (UTR) of the GOI, but downstream of the promoter (Figure 1). When the GOI is transcribed, the siRNA is co-transcribed. Three days after transfection, a messenger RNA (mRNA) is transfected into the cells, coding for a selection marker. In this study, we chose the CD4 mRNA, but fluorescent proteins are another option. In this system, if the GOI is transcribed, the siRNA is also transcribed, which silences the production of CD4 (Figure 2). Thus, productive clones can be selected from nonproductive clones using magnetic bead selection with antibodies against CD4, a standard immunological process. Alternatively, cells could be selected based upon expression, or lack thereof, of a fluorescent protein using fluorescence-activated cell sorting (FACS). Importantly, because the selection marker is introduced as an mRNA rather than as a DNA molecule, even in the absence of the siRNA, the molecule will be degraded over the course of days, eliminating the burden of producing the selection marker. This transfection and selection process can be repeated over multiple cycles until the desired purity is obtained. Using this approach, we were able to obtain a stable pool in 10 days.2
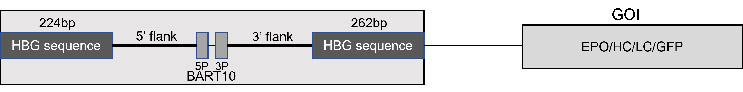
Figure 1: Schematic of the siRNA construct upstream of the gene of interest.

Figure 2: Overview of the selection process. When the GOI is transcribed, the siRNA is also produced. When the CD4 mRNA is introduced, it is silenced by the siRNA allowing for selection based upon the presence of CD4 protein on the surface using magnetic beads or fluorescent antibodies.
Transposon-based Technologies
Transposons are mobile genetic elements that can insert in different locations within the chromosome and move from one location to another. In the transposon system, the genes of interest are cloned into a vector with inverted terminal repeats (ITRs) at each end of the sequence (Figure 3). Transposases, enzymes that recognize specific target sequence within the ITRs known as directed repeats (DRs), bind to the transposon, cut the chromosomal DNA at the transposon recognition sites, and insert the vector DNA. Ligation occurs by the non-homologous end joining (NHEJ) pathway. This provides an opportunity for tens of copies of the GOI to be integrated into the host DNA. Typically, heterologous and/or engineered transposons are used, such as the engineered Tc1/mariner transposon “Sleeping Beauty,” the insect-derived “PiggyBac” system, and the “Leap-In Transposase” system, derived from a frog transposon. The transposase enzymes can be introduced into the cell on plasmid vectors separate from the transposon or as mRNAs or recombinant proteins to prevent stable incorporation of the transposase. A notable advantage of the transposon system is that, unlike plasmid incorporation, no bacterial DNA sequences are inserted into the chromosome, reducing the risk of silencing.3,4

Figure 3: Example of a synthetic transposon, showing the insertion region (blue), the inverted terminal repeats (red) and the transposase (black) facilitating insertion into the genome.
A variety of transposons have been employed in mammalian cell lines, particularly CHO cells to produce recombinant proteins with higher incorporation efficiencies and shorter timelines (e.g., from transfection to GMP-grade manufacturing in under three months). In addition, transposons are more likely to incorporate into transcriptionally active sites in the chromatin than with random integration. Studies have also shown expression of multiple transgenes and delivery of large payloads (i.e., > 100-kb of DNA). The Leap-In Transposase system demonstrated very good pool homogeneity and titers from 2.0 to 5.0 g/L without generating clonal cell lines, permitting the generation of preclinical and clinical material much more rapidly. Increases in product titers can be quite significant with some studies reporting up to 25-fold increases in titer from transposon-derived clones compared with conventionally derived clones.

Targeted Integration
As described above, the current state-of-the-art approach to generating cell clones relies on random integration of the GOI into the chromosomes, metabolic or antibiotic selection, followed by extensive screening of clones to identify high productivity clones. An alternative is the use of targeted integration in which the GOI is specifically targeted to a highly active transcriptional site or “hot spot.” These hot spots are generally identified by random integration of a reporter gene such as a fluorescent protein (e.g., green fluorescent protein [GFP], mCherry), followed by FACS sorting, selection, and cloning of highly fluorescent cells. Using a site-specific recombinase (e.g., Cre, Flp, Bxb1), recombinase-mediated cassette exchange (RMCE) occurs, excising the fluorescent protein and replacing it with the GOI (Figure 4a). The integration site is often referred to as a universal landing pad. More recently, programmable nucleases such as CRISPR/Cas9 have been applied for targeted integration, potentially allowing insertion into any chromosomal locus (Figure 4b). These approaches create highly reproducible systems, into which any GOI can be cloned. As in the case of transposons, these approaches also remove bacterial DNA sequences from the insertion sequence and allow for regulation of insert direction and genome–transgene junctions.
Figure 4a

Figure 4b
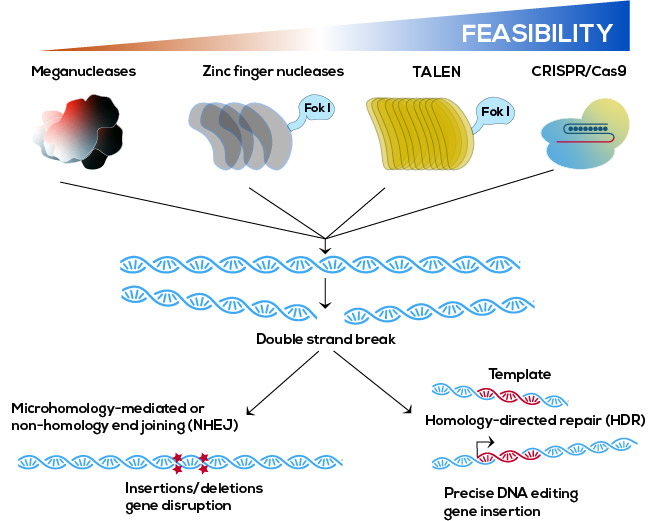
Figure 4: a. Recombinase-mediated cassette exchange. The recombinase catalyzes the exchange of the gene of interest into the landing pad based on the presence of the recombinase recognition sites on both the landing pad and the insertion vector. b. Examples of programable nucleases that can be used to insert a gene of interest into a specific chromosomal site.
In the case of programable nucleases, the desired integration site must be identified, generally by bioinfomatic or systems biology approaches, though some specific sites have been previously identified.6 More recently, RCME has been combined with nuclease mediated site-specific integration to target the landing pad into a previously identified site, avoiding the random integration and screening of the landing pad. Newer approaches have also included creating multiple landing pads to increase gene copy number or inserting tandem repeats of the GOI. One ongoing challenge is the low efficiency of homology directed targeted integration efficiency, particularly in CHO cells. While CRISPR/Cas9 editing is more effective than earlier types of nucleases (e.g., ZFN, TALEN), the overall efficiency of incorporation is still low. As the technologies improve, we can expect that the incorporation efficiency in CHO cells will improve as well.5,6
Conclusions And Perspectives
Biologics should continue to dominate the novel therapeutics introduced annually, particularly for high-value pharmaceuticals. For multibillion-dollar-a-year drugs, millions of dollars are lost for every day the drug is not on the market. This financial drive combined with a desire to treat conditions with unmet clinical needs increases the impetus to accelerate cell line development. In addition, the introduction of difficult to express proteins such as bispecific antibodies will require more exacting cell line development processes, creating additional demands for these novel technologies.
Acknowledgements:
- Figures 1 and 2 reprinted from Journal of Biotechnology, Vol. 325, V. Muralidharan-Chari et al, PTSelect™: A post-transcriptional technology that enables rapid establishment of stable CHO cell lines and surveillance of clonal variation pp:360-371. Copyright (2021) with permission from Elsevier.
- Figure 3 provided courtesy of ATUM.
References:
- V. Tejwani, M. Chaudari, T. Rai, and S.T. Sharfstein, High-throughput and automation advances for accelerating single-cell cloning, monoclonality and early phase clone screening steps in mammalian cell line development for biologics production, Biotechnology Progress, Sep 3;e3208 (2021) doi: 10.1002/btpr.3208
- V. Muralidharan-Chari, Z. Wurz, F. Doyle, M. Henry, A. Diendorfer, S.A. Tenenbaum, N. Borth, E. Eveleth, S.T. Sharfstein, PTSelect™: A post-transcriptional technology that enables rapid establishment of stable CHO cell lines and surveillance of clonal variation, Journal of Biotechnology, 325:360-371 (2021) doi: 10.1016/j.jbiotec.2020.09.025
- M. Wei, C.L. Mi, C.Q. Jing, T.Y. Wang, Progress of Transposon Vector System for Production of Recombinant Therapeutic Proteins in Mammalian Cells, Frontiers in Bioengineering and Biotechnology, 10: 879222 (2022) doi: 10.3389/fbioe.2022.879222
- N. Sandoval-Villegas, W. Nurieva, M. Amberger, Z. Ivics, Contemporary Transposon Tools: A Review and Guide through Mechanisms and Applications of Sleeping Beauty, piggyBac and Tol2 for Genome Engineering, International Journal of Molecular Sciences, 22 (10): 5084 (2021) doi: 10.3390/ijms22105084
- J-S. Lee, H. Faustrup Kildegaard, N.E. Lewis, G-M. Lee, Mitigating Clonal Variation in Recombinant Mammalian Cell Lines, Trends in Biotechnology, 37 (9): 931-942 (2019) doi: 10.1016/j.tibtech.2019.02.007
- S.W. Shin, J.S. Lee, CHO Cell Line Development and Engineering via Site-specific Integration: Challenges and Opportunities. Biotechnology and Bioprocess Engineering, 25: 633–645 (2020) doi: 10.1007/s12257-020-0093-7
About the Author:

Susan Sharfstein, Ph.D., is a professor at the University at Albany’s College of Nanotechnology, Science, and Engineering. She leads the Sharfstein laboratory, which is focused on the role of culture conditions and cell physiology and the use of living systems for industrially relevant processes. Her lab is developing new tools to better understand cultured cells and bioprocesses. She is a widely published author and researcher who has led and collaborated on dozens of peer-reviewed works. In the past, she served as a SUNY Research Foundation faculty fellow, and she was a biotechnology subject editor for the Elsevier Life Science Reference Module. She was a 2017-18 Fulbright Global Scholar honoree.