
Introduction To The ASTM E3106 "Standard Guide To Science-Based And Risk-Based Cleaning Process Development and Validation"
By Andrew Walsh, Thomas Altmann, Joel Bercu, Ph.D., Alfredo Canhoto, Ph.D., David G. Dolan Ph.D., Andreas Flueckiger, M.D., Igor Gorsky, Jessica Graham, Ph.D., Ester Lovsin Barle, Ph.D., Ovais Mohammad, Mariann Neverovitch, and Osamu Shirokizawa
Part of the Cleaning Validation For The 21st Century series
This article will provide a detailed discussion of the science-, risk-, and statistics-based approaches in the American Society for Testing and Material (ASTM) E3106 Standard Guide for Science Based and Risk Based Cleaning Process Development and Validation.
As discussed in the first article of this series,1 many initiatives from regulatory agencies and the industry have directed the industry toward more science-, risk-, and statistics-based approaches for cleaning validation. The more important milestones for cleaning validation include the Barr Labs Court Case,2 the 1996 revisions to the GMPs,3 and the introduction of the ISPE Risk-MaPP Guideline4 in response to the 1996 revisions.
In 1996, the FDA proposed revisions to the GMPs that stated:
"FDA expects manufacturers to identify any drugs that they produce that present the risk of cross-contamination and to implement measures necessary to eliminate that risk."
Around the same time, some regulators and industry subject matter experts had begun to challenge the legitimacy of the 0.001 of a dose, or 10 ppm, for setting acceptance limits for cleaning validation.5 It was becoming recognized that limits calculated using these criteria were widely variable and could lead to low-risk products unnecessarily requiring dedicated facilities. In some cases, these traditional limits were not set low enough. More importantly, it was also becoming understood these limits did not consider all of the available toxicological and clinical data for drug products in determining cleaning validation limits.
A team was formed in 2004 that would eventually write the Risk-MaPP Guide. In order to address the challenge of the 1996 revisions, a new approach to setting cleaning validation limits was required that considered all of the available toxicological and clinical data for drug products. A well-established approach that was used in determining Occupational Exposure Limits (OELs) for pharmaceutical worker exposure to drugs was selected as the model. The approach for OELs used the principles of calculating Acceptable Daily Intakes (ADIs) to calculate an OEL for a pharmaceutical worker over an 8-hour day. However, the ADI itself was closely associated with food and oral exposure and the FDA objected to the use of this term. Although the calculation remained the same, the name was changed to Acceptable Daily Exposure (ADE), which the FDA found acceptable. Risk-MaPP defined the ADE as:
"A dose that is unlikely to cause an adverse effect if an individual is exposed, by any route, at or below this dose every day for a lifetime."
It should be obvious that this definition — an exposure level, every day, for a lifetime, that would have no adverse effect on a patient by any route of exposure — is very stringent. Actually, this is extremely stringent. Since the ADE is based on all the available toxicological and clinical data for the compound, it can be used for setting appropriate health-based limits not only for worker exposure but for cleaning as well. So, the ADE was an obvious replacement for the 10 ppm and 0.001 therapeutic dose criteria originally introduced by Eli Lilly.
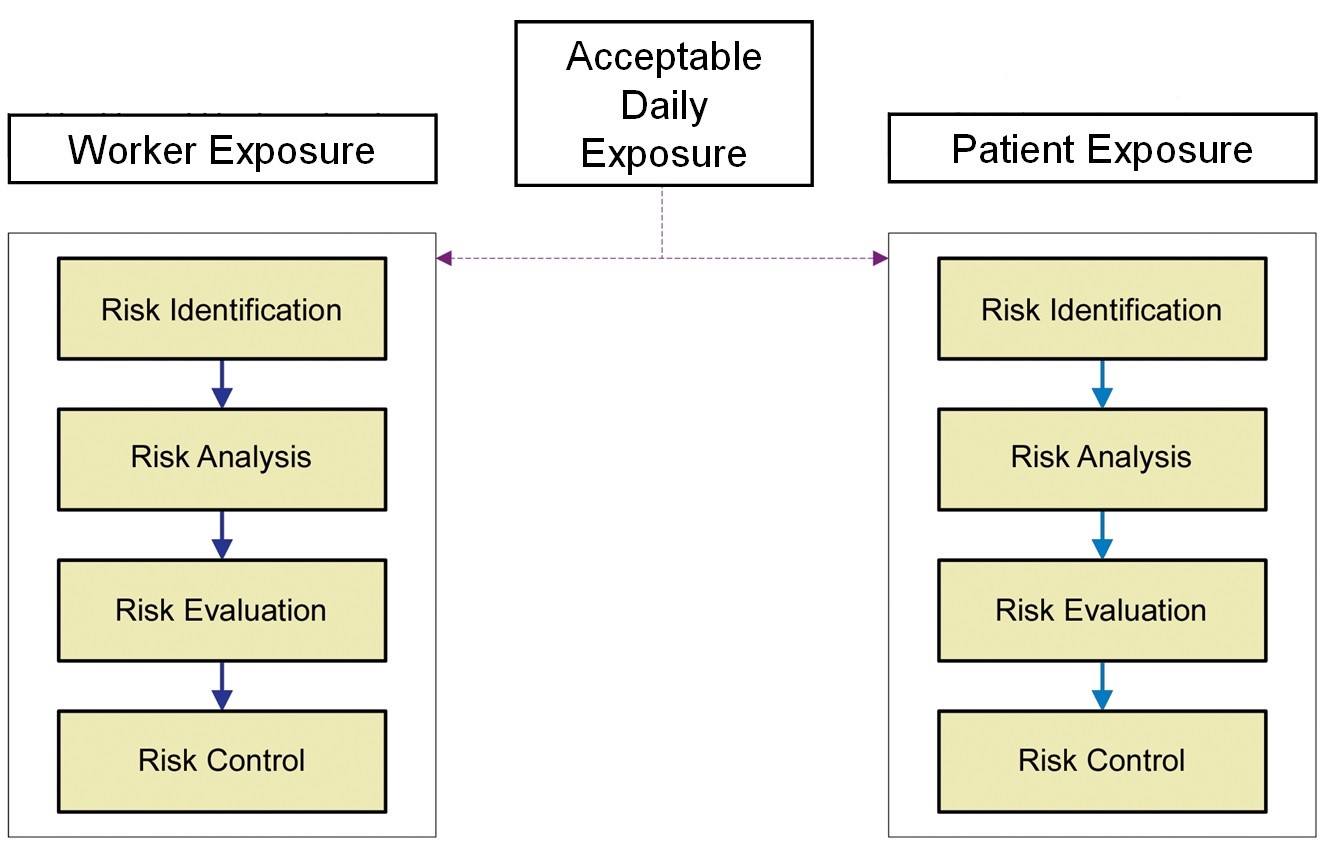
Figure 1: Overview of the Risk-MaPP process
Figure 1 shows an overview of the Risk-MaPP process. The bases for acceptable worker exposure and acceptable patient exposure were now the same — the ADE. The approach that had been used for many years to address worker exposure (i.e., occupational exposure limits) would also be used to address patient exposure. While an appropriate basis had now been decided for risk identification, there still needed to be a way to work down the column on the right hand side for acceptable patient exposure to determine the risk analysis, risk evaluation, and risk control.
ASTM E3106
A global multidisciplinary team was formed starting in 2007 with representatives from the U.S., Europe, and Asia to develop a science-based and risk-based approach for the right-hand column in Figure 1 to address patient exposure from residues after cleaning. All candidates for the team had to agree to three principles:
- Science-, risk-, and statistics-based standard: The standard would reflect the new directions that regulators and industry had been pointing toward. That is, science-based, risk-based, and statistically based approaches to cleaning process development and cleaning validation. Concepts and principles, such as those found in ICH Q9, the FDA’s 2008 draft process validation guidance,6 and others, would provide the philosophy.
- Embrace new ideas: The standard would step into the future and be an improvement over current industry and regulatory practices. The standard would propose new thinking and procedures that may not be present at the team members' current employers. There would be very few companies that would not have to change something. This standard should be new and bring new value.
- Let go of the past: Team members needed to embrace the points above and not try to protect any practices that may be in place at their companies. All team members had to be willing to advocate for change in the industry and even within their own companies.
These three principles proved to be too difficult for some candidates, but a team was eventually selected with members who had extensive experience in cleaning and validation and expertise in one or more areas. The team also had representation from pharmaceutical, biotech, API, and clinical manufacturing (Table 1) companies.
Table 1: ASTM E3106 Standard Guide Team Members
E3106 Principles From ICH Q9
There are two primary principles of quality risk management found in ICH Q9 that were fundamental in developing ASTM E3106. These are:
- The evaluation of the risk to quality should be based on scientific knowledge and ultimately link to the protection of the patient; and
- The level of effort, formality, and documentation of the quality risk management process should be commensurate with the level of risk.
It is also very important to also recognize that that these two primary principles are applicable to validation (Annex II.6). More significantly, ICH Q9 also states that they are applicable to cleaning, including setting acceptance limits for cleaning processes (Annex II.4).
So, we can reword the ICH Q9 principles to cover cleaning as follows:
- “The evaluation of the risk in cleaning should be based on scientific knowledge and ultimately link to the protection of the patient; and
- The level of effort, formality, and documentation of the cleaning validation process should be commensurate with the level of risk."
ICH Q9 also offered a framework for implementing a quality risk management process upon which Risk-MaPP was structured (Figure 2). The ASTM E3106 standard was also structured on the ICH Q9 framework and the standard sections step through the ICH Q9 process.
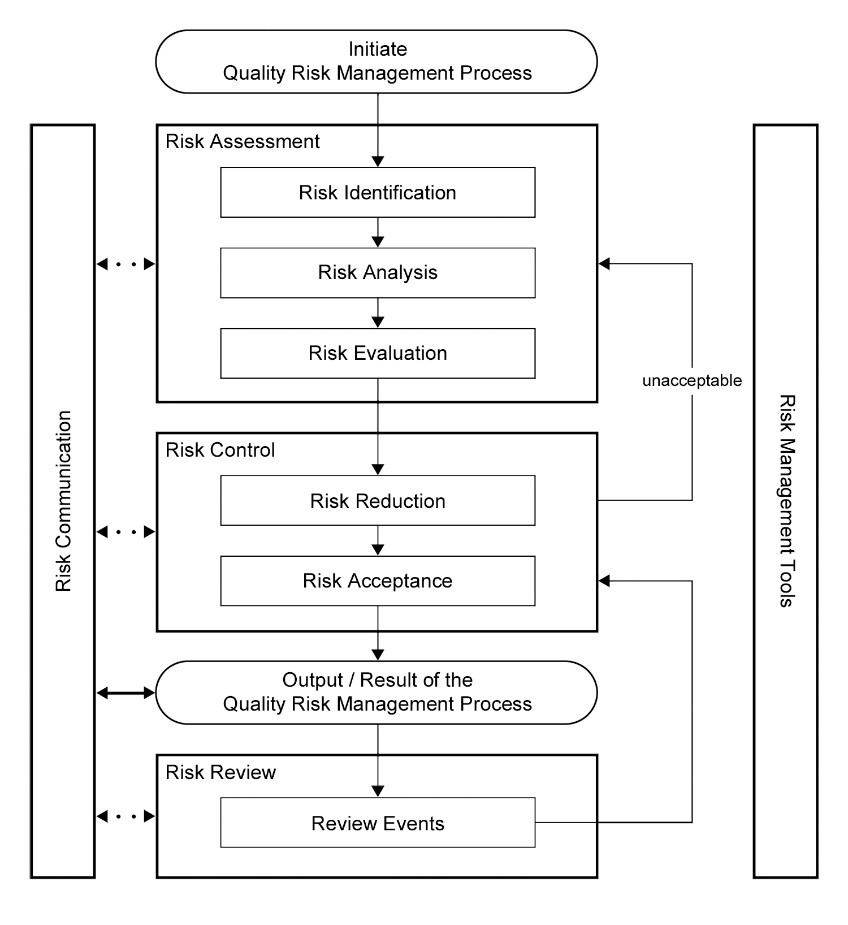
Figure 2: Overview of a typical quality risk management process
Concepts From FDA Process Validation Guidance
In 2008, the FDA released a draft of its updated process validation guidance6 to align with the product life cycle concept and with existing FDA guidance on ICH Q8-Q10. The final guidance was published in 2011 and described concepts that are directly applicable to cleaning and cleaning validation. The FDA has also stated publicly that the new guidance applies to cleaning validation.7 We can simply add "cleaning" to the elements of the process validation guidance as shown below.
- Cleaning Process Design – Building and Capturing Process Knowledge and Understanding
- Application of design of experiment to cleaning
- Multifactorial interactions
- Using risk analysis tools to screen potential variables
- Cleaning Process Qualification
- Use of statistical methods in analyzing all collected cleaning data
- Continued Cleaning Process Verification
- Use of statistical process control techniques
- Continuous Improvement
- Use of historical data (monitoring, etc.) or technological advances for improvement of cleaning processes
The concepts in the process validation guideline can be easily worked into a framework for a science-, risk-, and statistics-based approach to cleaning.
Combining the ICH Q9 Risk Management approach with the FDA's Process Validation Guidance provides an excellent means to implement the Risk-MaPP approach and address the 1996 proposed revisions for making decisions on whether to manufacture a product in a dedicated facility or on dedicated equipment. The combination of these two approaches became the ASTM E3106 Standard Guide.
The ASTM E3106 Cleaning Risk Management Process
In ICH Q9, risk assessment is broken down into three stages: risk identification, risk analysis, and risk evaluation. Before starting to work through a risk assessment, it is necessary to understand what is meant by the word "risk."
In ICH Q9, risk is defined as:
Risk = f (Severity of a Hazard, Exposure to the Hazard, Detectability of the Hazard)
For the purposes of cleaning, risk can be further defined as a function of the toxicity of of cleaning residues, the likelihood and level of cleaning residues that may be present, and the detectability of these cleaning residues:
Risk = f (Toxicity of a Residue, Level of the Residue, Detectability of the Residue)
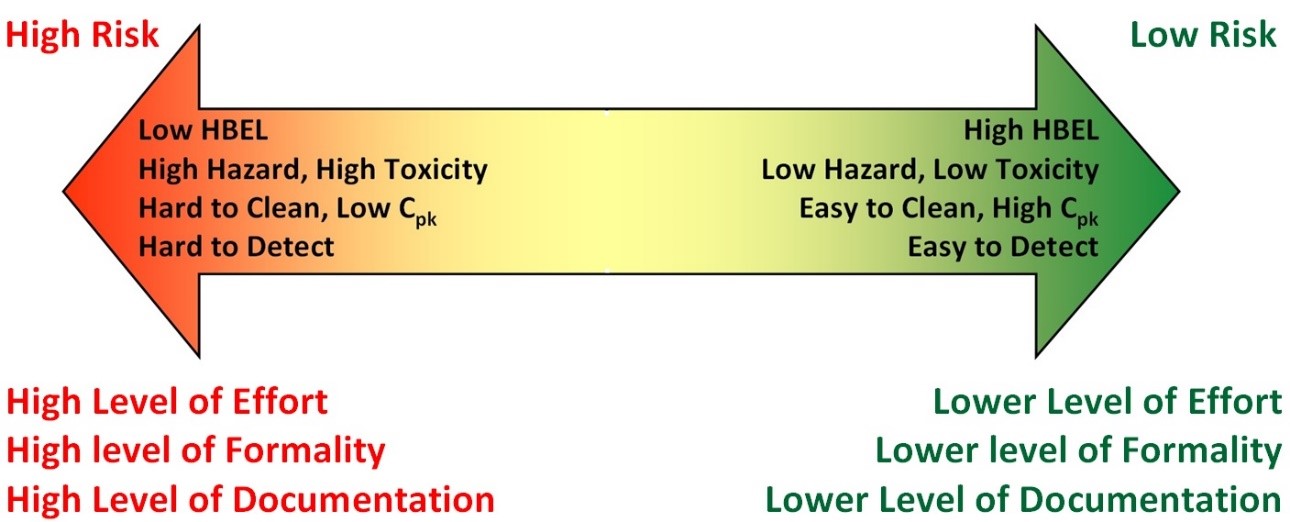
Figure 3: Cleaning risk continuum using ICH Q9 principles — this is a visual depiction of the continuum of these factors and how they impact risk. Note: HBEL is the Health Based Exposure Limit, which is equivalent to the ADE or PDE.
E3106 is based on ICH Q9’s risk principle that the "level of effort, formality, and documentation of the validation should be commensurate with the level of risk." This provides a means to protect patient safety and determine effective and efficient cleaning validation activities that would focus efforts and resources where they provide the most value. An overview of the Quality Risk Management process in E3106 is shown in Figure 4.
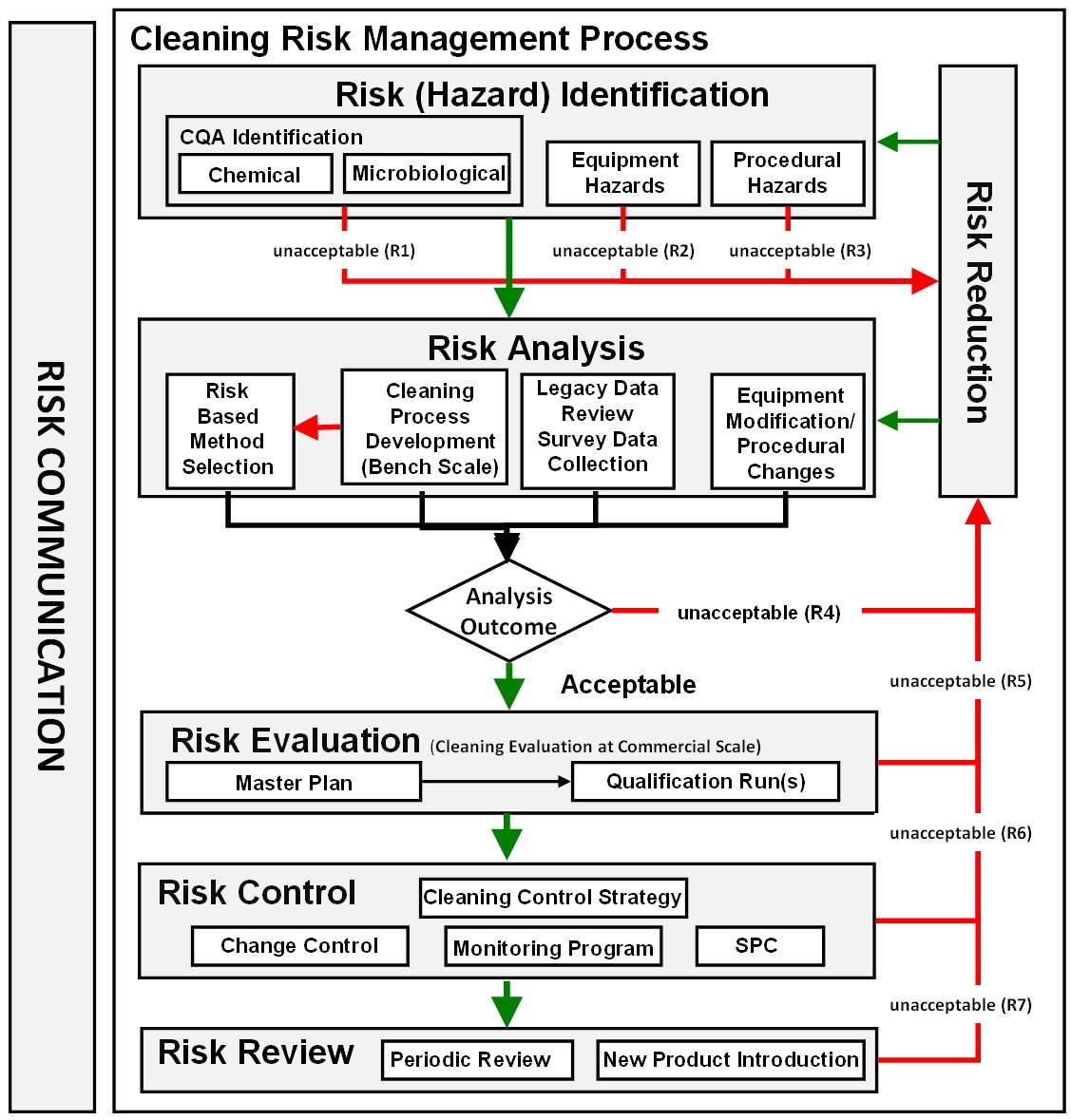
Figure 4: Overview of the ASTM E3106 cleaning risk management process — this diagram is a compilation of all the steps in the cleaning QRM process in E3106 that are described in more detail in the sections below
In order to do this, it is required that we identify the hazards, measure the patient exposure to the hazard, and be able to detect the hazard. The first step in this process is to identify the hazards.
Risk (Hazard) Identification
In ASTM E3106, risk identification encompasses not only the identification of the hazards from process residues but also any hazards associated with equipment design and any hazards associated with the cleaning procedures (SOPs). See Figure 5.
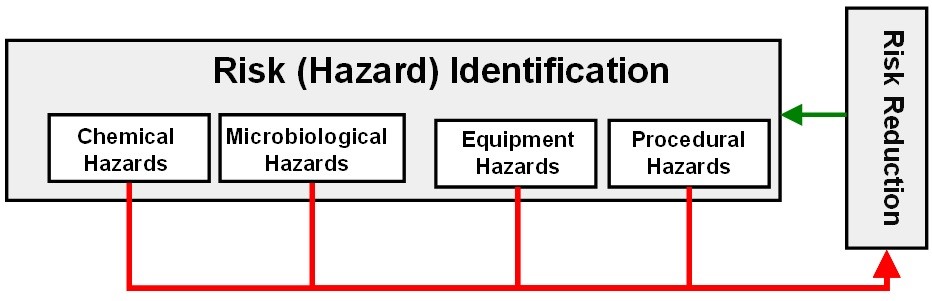
Figure 5: Four hazards identification
The potential hazard of a cleaning residue should be determined by a qualified expert. E3106 utilizes the HBEL for evaluating the risk to patients from the cleaning of manufacturing equipment and devices and will be aligned with the recently approved E3219 standard. A toxicity scale has been developed (based on the HBEL) that allows the toxicity of compounds to be easily visualized so comparisons can be easily made of the hazards (Figure 6).8
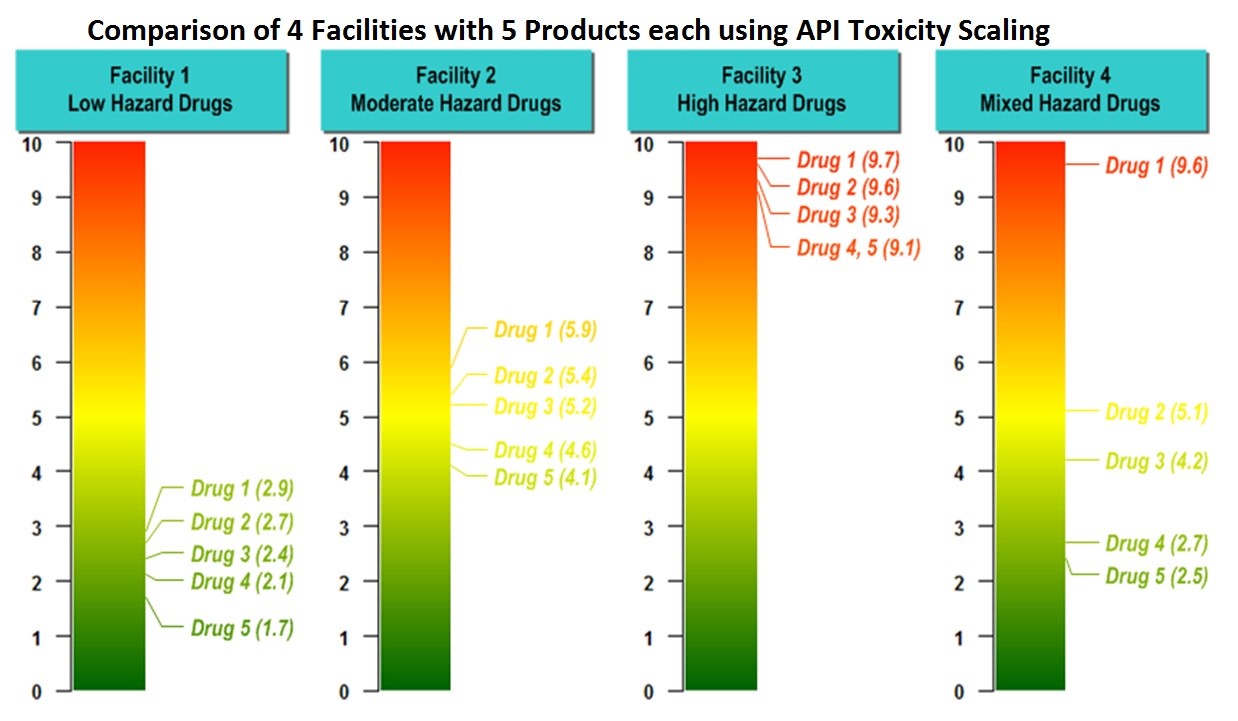
Figure 6: Example comparison of toxicity scores for chemical hazards for different facilities
Potential microbial hazards from a previous product and the possibility of proliferation after a cleaning process need to be identified.
The potential hazards presented by equipment design need to be examined, such as difficulty in cleaning or possible product residue buildup. Equipment should be designed to facilitate cleaning, inspection, and monitoring and should be modified or replaced if cleaning issues cannot be resolved. This may be especially relevant for older equipment that has been in operation for a long time but was not designed to be cleaned, inspected, and monitored effectively.
Before use, cleaning procedures should be subjected to risk assessments through a cleaning FMEA or other risk management tool, to eliminate or minimize the risk of a failure of the cleaning process9 (e.g., poka-yoke, ポカヨケ), improve the cleaning procedures, and make the cleaning procedures more reliable and robust.
Even though this is early in the risk assessment process, risk reduction steps should be taken if a risk (hazard) is found.
Risk Analysis:
After identifying the hazards posed, the risks associated with them should be analyzed. This risk analysis should involve the cleaning process development, facility/equipment design review, cleaning procedure review, and the review on selection of analytical methods. The analysis should also determine what steps can be taken to reduce or eliminate any identified risks (Figure 7).
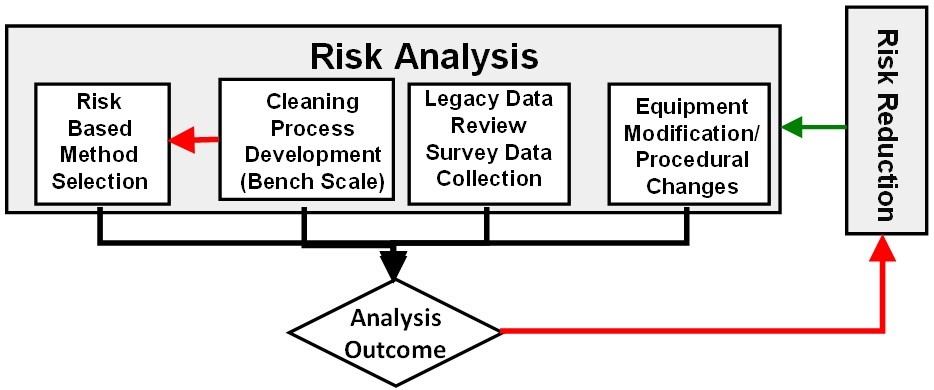
Figure 7: Cleaning risk analysis
Cleaning Process Development
Cleaning processes should be developed with an understanding of the hazard associated with the residues and to determine the appropriate cleaning process parameters needed to reduce residue levels as low as practical. The output of the cleaning process development should be used to create the cleaning standard operating procedures (SOPs). Laboratory scale or “bench-scale” studies can provide valuable sources of cleaning process knowledge and cleaning process understanding (Figure 8). Studies can also be conducted in small-scale equipment designed to simulate the actual manufacturing equipment and conditions.9

Figure 8: Automated high-throughput cleanability testing device (patent pending) – this device is microprocessor controlled, running Windows 10 IoT Core, and can run multiple programs while independently controlling and monitoring up to nine test stations. Test coupons are attached to one of the nine test stations.
Risk-Based Analytical Method Selection
The choice of analytical methods should be science- and risk-based. The simplest technique that is appropriate and can be justified should be selected as it reduces the potential for errors and unnecessary expenses. The hazard identification, combined with information from the cleaning process development, can be used to justify the method selection. Typically, as the level of risk increases, the level of analytical sophistication should increase along with it (Figure 9).
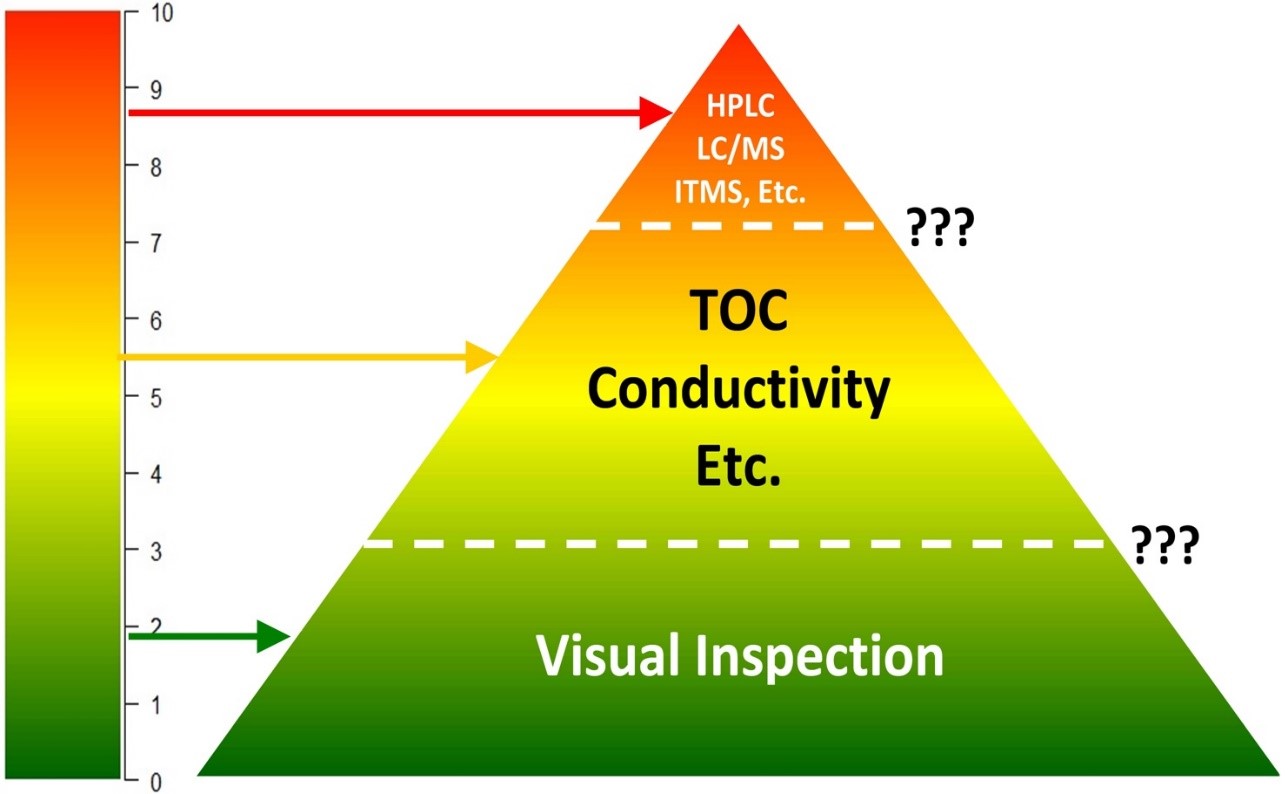
Figure 9: Risk-based selection of analytical methods — companies will need to determine at what level of risk they will need to move from one methodology to another (See Figure 13).
Specific methods, such as high-performance liquid chromatography (HPLC), should be considered for high-hazard products or high-risk situations. Non-specific methods, such as total organic carbon (TOC), UV, conductivity, pH, and visual inspection detect the presence of multiple ingredients that can be acceptable in lower-risk situations. Using visual inspection alone for validation may be acceptable,11 but only when a risk assessment has shown that the risk is low and 100 percent of the equipment surface can be inspected under appropriate viewing conditions. Two detectability scales (Figure 10) have been developed, one for TOC (or any method) and the other for visual inspection, that can be used to easily visualize how well these methods can be relied upon.12,13
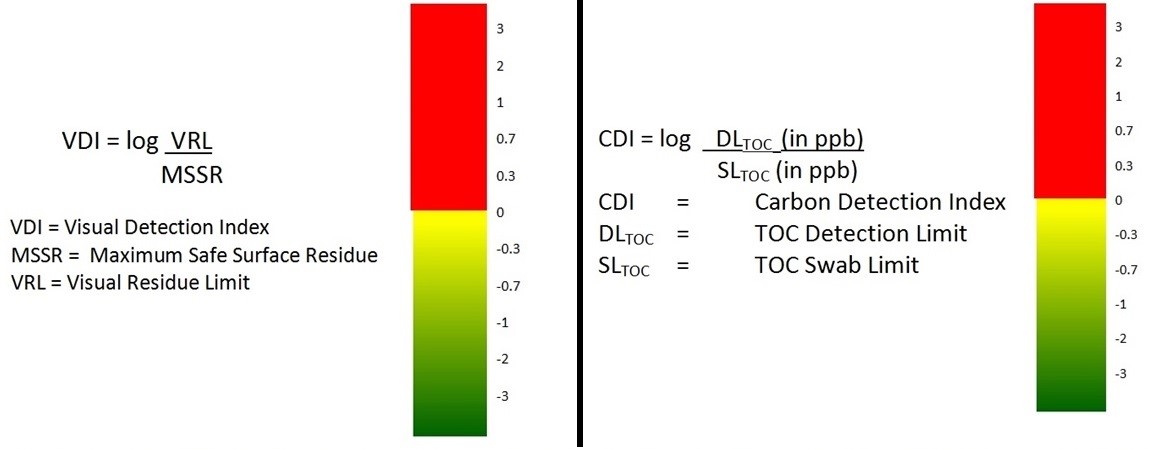
Figure 10: Detectability scales for analytical methods — scales for any analytical method can be obtained by simply taking the logarithm of the ratio of the detection limit divided by the swab/rinse limit. These logarithmic scales equal 0 when the values of the swab/visual limit and detection limit are equal. They become negative when the detection limit is lower than the swab/visual limit and positive when it is higher.
Legacy Data Review and Collection of Survey Data – The history of cleanings, along with any deviations, investigations, and corrective actions, should be reviewed. All cleaning data should be analyzed statistically. These reviews provide understanding and knowledge about the cleaning process and can provide useful information in a risk analysis. These reviews may help identify cleaning process parameters to be used in cleaning process development studies and determine the likelihood of a cleaning failure. Sufficient historic data should be available to analyze and draw conclusions. If sufficient historical data is not available, then cleaning data from current cleaning processes should be collected for analysis.
Equipment Modification/Procedural Changes – Equipment design has an impact on its cleanability and should be considered as part of the risk analysis. Where satisfactory cleaning results cannot be achieved because of limitations in the equipment design or continued use of overly conservative traditional limits, the equipment may need to be modified, replaced, or dedicated. SOPs should be reviewed and, if necessary, modified to prevent potential failures due to equipment design.11
Risk Acceptance
After all risk reductions from the hazard identification and risk analysis steps have been completed, it is at this point that the residual risk is either accepted, and the cleaning risk management process can move on to the risk evaluation step, or the risk is rejected and further reduction efforts are needed (Figure 11).
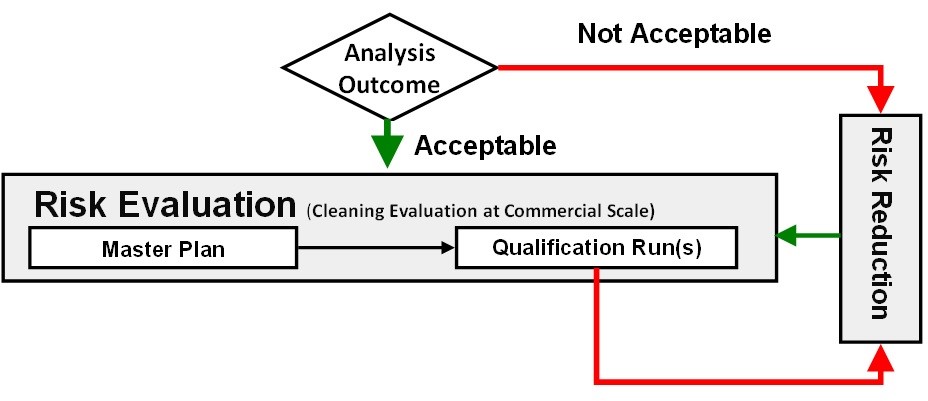
Figure 11: Cleaning risk acceptance and risk evaluation
Risk Evaluation:
Information from the risk (hazard) identification and risk analysis steps is used to determine what cleaning qualifications studies are necessary, how many studies are necessary, and to identify appropriate risk control mechanisms.
The master plan for cleaning should be developed after the hazard identification of the process residues, the review of legacy data/survey data, and the review of equipment design and cleaning procedures are found to be acceptable (Figure 10). The combination of hazard identification and the risk analysis provide the basis to develop the master plan for cleaning and select the cleaning control strategy. At this point in the cleaning risk management process, if the hazard identification and the risk analysis have been done adequately, the qualification runs should only be confirmations and there should be no failures and no need for risk reduction efforts.
The recommended approach for evaluating cleaning data (in terms of surface residue based on the HBEL) collected during qualification runs is to compare the cleaning data to the maximum safe surface residue (MSSR) based on the HBEL for that process residue and determine the process capability.14 The comparison of cleaning data to MSSRs can demonstrate whether process residues on equipment product contact surfaces pose a significant risk to patients and can demonstrate what the margin of safety is for that process residue. A narrow margin of safety is an indication of higher risk and vice versa. A process capability scale15 has been developed that allows the process capability of cleaning processes to be easily visualized so comparisons can be easily made of the effectiveness of cleaning processes (Figure 12).
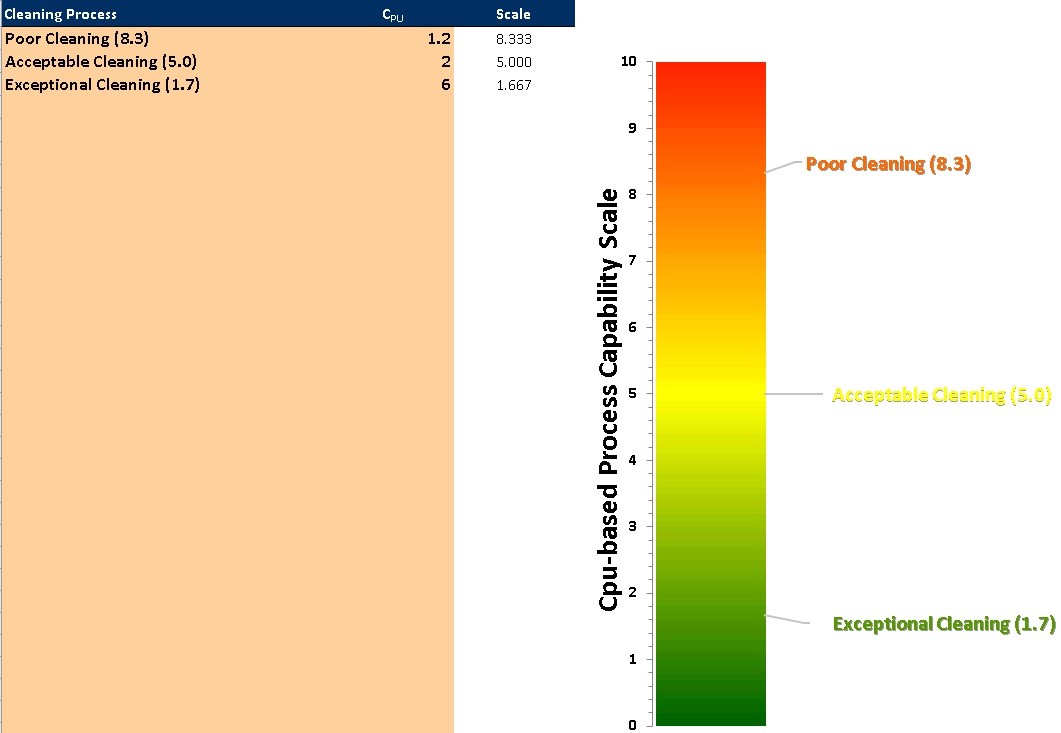
Figure 12: Example of process capability scores for chemical hazards
A similar procedure can be used for assessing microbial risk.16, 17
The results of all cleaning runs should be collected in a database so that the HBELs of new products can be evaluated against the existing cleaning processes to assess the potential capability of the cleaning process to reduce residues of the new product. This can be a simple exercise of recalculating the process capability for the current data using the limit from the new HBEL. This assumes that the same level of process residues is expected for the new product. A cleanability study as described above can quickly confirm this.
If the potential process capability for a new product is determined to be too low, this could result in the need for further cleaning process development studies to ensure that the cleaning procedure can affectively remove the new product to safer and more acceptable levels.
Risk Control
A cleaning control strategy should be developed based on the data collected during cleaning process development and during the qualification runs after any risk reduction activities are completed (Figure 13).

Figure 13: Cleaning risk control and risk review
The cleaning control strategy may include continued monitoring of critical cleaning process parameters and sampling and testing of critical cleaning quality attributes in high-risk situations or may be only visual inspection in low-risk situations.
Data that have been collected in the risk evaluation or risk control stages should have Statistical Process Control (SPC) limits obtained from the analyses of these data. The SPC limit should then be used to monitor the cleaning processes and, in place of the MSSR limits used for the risk analysis in risk evaluation. A matrix18 based on the toxicity of compounds and the capability of their cleaning processes has been developed that provides a means of selecting a control strategy based on ICH Q9 principles (Figure 14).
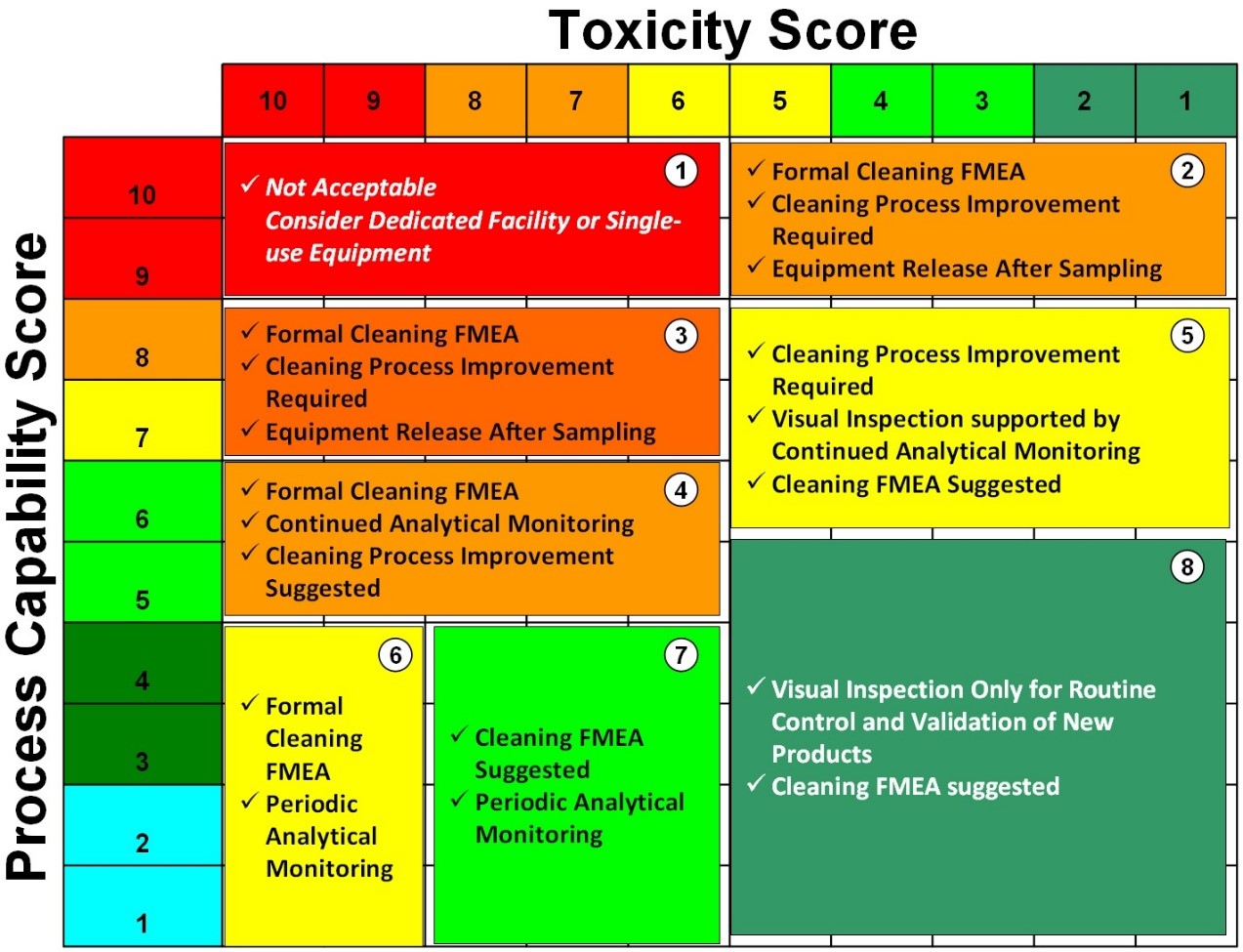
Figure 14: Example of Shirokizawa Matrix for level of effort, formality, and documentation
Risk Review
If the hazard identification, risk analysis, and risk reduction steps have been adequately performed, then the risk review step should be uneventful.
The only significant event in risk review should occur when a new product is being introduced. Before introducing a new product to shared manufacturing equipment or a facility, a decision should be made and documented about the appropriateness of manufacturing this product in the equipment or facility. This would include an evaluation of all relevant toxicological data by a qualified expert to determine an HBEL of the new product. The process capability of the existing cleaning processes should be evaluated to determine the potential effectiveness of the cleaning processes for removing residues of the new product. At this point, if the new product is acceptable and the cleaning processes appear capable, the cleanability of the product should be determined by laboratory-scale testing to confirm that the cleaning process parameters are still appropriate. If the new product is not cleanable with the existing cleaning process, cleaning process development would be necessary, including qualification studies for the new cleaning process. Otherwise, no additional cleaning process development or qualification studies would be necessary.
Dashboards are widely used in business to provide simple "at-a-glance" tools that can quickly show visual representations of complex relationships among business metrics, key performance indicators (KPIs), or any other data important to making decisions about a business process. Dashboards communicate knowledge efficiently and simplify the decision-making process by making multiple sources of data and their relationships easy to visualize. Ultimately, a critically important process such as quality risk management would benefit from a dashboard that could easily present the multiple sources of data so that decisions concerning risk could be made efficiently and with confidence.
The scales discussed in this article can be used to develop such a dashboard. Figure 15 shows an example of how new compounds can be quickly and easily evaluated to determine whether the current cleaning process and analytical methods allow these compounds to be manufactured in a shared equipment facility. Cleaning limits (e.g., MSSRs) based on the new products’ HBELs are determined and evaluated using the facility's existing cleaning data and estimates of the potential cleaning process capability against the limit for the new product. The new cleaning limits can be compared to the known detection limits (e.g., TOC, visual inspection) using the detectability scales12,13 to determine if the existing methods are capable of acceptably detecting the new products.
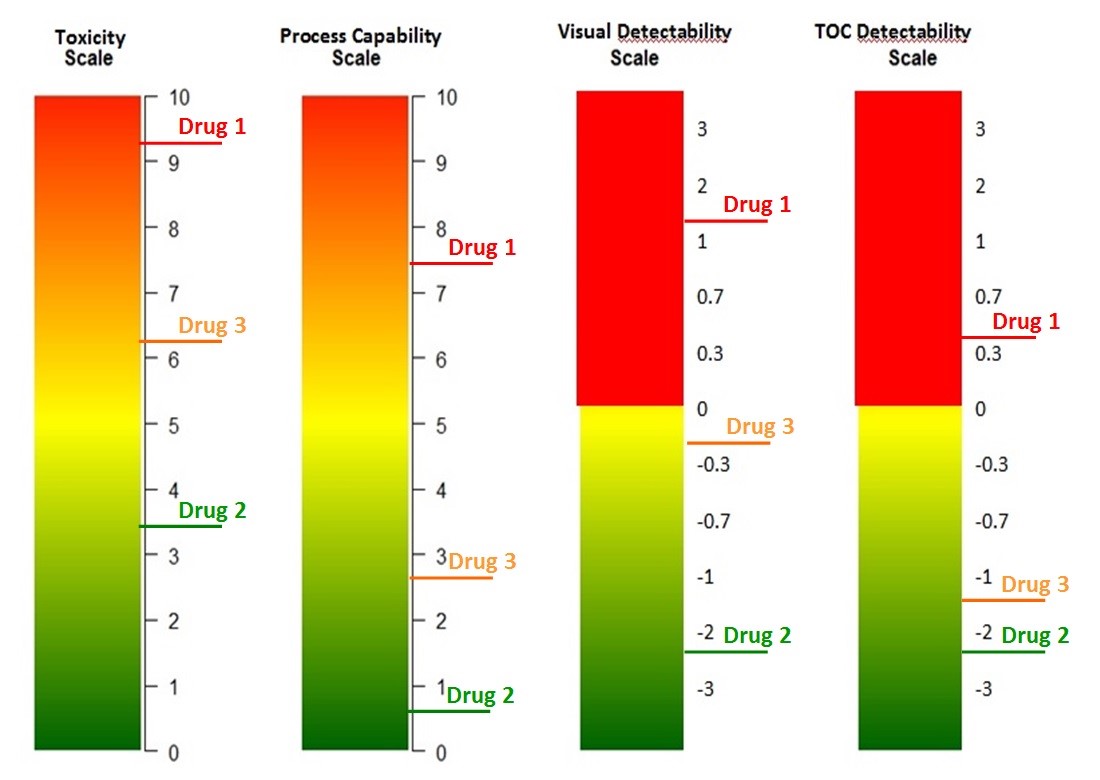
Figure 15: Example of cleaning risk dashboard
Immediately, it can be seen that Drug 1 is a very toxic compound and that the current cleaning process cannot adequately clean it to prevent cross contamination issues. (Note: Process Capability can be evaluated based on existing cleaning data compared to the limits required by the new compound.) In addition, residues cannot be detected at a safe level, visually or even by TOC. Introducing this drug would require substantial improvements in both the cleaning process and in analytical methodologies. Most likely, a manufacturer would need to dedicate equipment or an entire facility to the manufacture of this drug.
Drug 2, on the other hand, is not highly toxic, and the current cleaning process can easily clean it to prevent cross contamination issues and any residues can be easily detected visually or by TOC. Introducing this drug would not require any improvements and initial manufacturing could possibly be evaluated by visual inspection only.
Drug 3 is somewhat toxic, but the current cleaning process could adequately clean it to prevent cross contamination issues and while residues cannot be detected visually, the TOC method is acceptable for detection. Introducing this drug would also not require any improvements.
Spreadsheets (created by Ovais Mohammad) for generating all of these scales are freely available for use under the GNU General Public License at the links provided below.20-22.
Summary
The new ASTM E3106 focuses much more attention on applying science and risk at the risk identification and risk analysis stages, including cleaning process development, than has been done in the past. Figure 4 shows all the steps discussed above in one diagram. Appropriate efforts at these early stages can provide reductions in the level of effort, formality, and documentation of the overall validation process, allow the selection of risk-based analytical methods (including visual inspection), and simplify the introduction of new products in the product matrix.
ASTM E3106 can be utilized to create new approaches to cleaning and cleaning validation that are based on science, risk, and statistics. By implementing a truly science-based approach, such as the use of the HBELs for risk analysis, with appropriate risk assessments, and with cleaning process development in place, a streamlined cleaning program may be readily developed that ensures patient safety and product quality while lightening the regulatory burden on industry.
The next article in this series will provide a detailed discussion of ASTM E3219, Standard Guide for Derivation of Health Based Exposure Limits (HBELs).
Peer Review
The authors wish to thank Bharat Agrawal, James Bergum, Ph.D., Sarra Boujelben, Gabriela Cruz, Ph.D., Parth Desai, Tri Chanh Nguyen, Miquel Romero Obon, Prakash Patel, Siegfried Schmitt, Ph.D., Basundhara Sthapit, Ph.D., and Joel Young for reviewing this article and for providing insightful comments and helpful suggestions.
References:
- Walsh A. and O. Shirokizawa, "Introduction to Science Based and Risk Based Cleaning Validation and the ASTM E3106 and E3219 Standard Guides". Pharm-Tech Japan, March 2020
- United States vs. Barr Laboratories, Inc. Civil Action No. 92-1744, U.S. District Court for the District of New Jersey: 812 F. Supp. 458. 1993 US Dist. Lexis 1932; 4 February 1993, as amended 30 March 1993.
- Current Good Manufacturing Practice: Proposed Amendment of Certain Requirements for Finished Pharmaceuticals. Federal Register / Vol. 61 No. 87 / Friday, May 3, 1996 / Proposed Rules
- ISPE Baseline® Guide: Risk-Based Manufacture of Pharmaceutical Products (Risk-MaPP), International Society for Pharmaceutical Engineering (ISPE), First Edition, September 2010.
- Walsh, Andrew, "Using TOC Analysis for Cleaning Validation" Barnett International Cleaning and Cleaning Validation Conference, Philadelphia, PA, February 2, 2001 DOI: 10.13140/RG.2.2.31546.64962
- FDA Guidance for Industry: Process Validation - General Principles and Practices January 2011, U.S. Food and Drug Administration (FDA), www.fda.gov.
- International Pharmaceutical Quality October 29, 2015 (Clarifying Questions Upfront is Key in Process Validation, US and EU PV Principles in Alignment — CDER’s McNally) https://www.ipqpubs.com/2015/10/29/clarifying-questions-upfront-is-key-in-process-validation-us-and-eu-pv-principles-in-alignment-cders-mcnally-stresses/
- Walsh, Andrew, Ester Lovsin Barle, Michel Crevoisier, David G. Dolan, Andreas Flueckiger, Mohammad Ovais, Osamu Shirokizawa, and Kelly Waldron. "An ADE-Derived Scale For Assessing Product Cross-Contamination Risk In Shared Facilities" Pharmaceutical Online, May 2017
- Song, Ruijin, Alfredo Canhoto, Ph.D., and Andrew Walsh, "Cleaning Process Development: Cleanability Testing and "Hardest-To-Clean" Pharmaceutical Products" Pharmaceutical Online, January 2019
- European Medicines Agency: Questions and answers on implementation of risk-based prevention of cross-contamination in production and “Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities," 19 April 2018 EMA/CHMP/CVMP/SWP/246844/2018
- Killilea, M.C., “Cleaning Validation: Viracept, 2007,” Journal of Validation Technology, November 2012, Vol. 18, Issue 4, www.ivtnetwork.com/jvt-journal.
- Walsh, Andrew, Thomas Altmann, Alfredo Canhoto, Ester Lovsin Barle, David G. Dolan, Andreas Flueckiger, M.D., Igor Gorsky, Robert Kowal, Mariann Neverovitch, Mohammad Ovais, Osamu Shirokizawa and Kelly Waldron. "A Swab Limit-Derived Scale For Assessing The Detectability Of Total Organic Carbon Analysis" Pharmaceutical Online, January 2018
- Walsh, Andrew, Thomas Altmann, Alfredo Canhoto, Ester Lovsin Barle, David G. Dolan, Mariann Neverovitch, Mohammad Ovais, Osamu Shirokizawa and Kelly Waldron. "An MSSR-derived Scale for Assessing the Detectability of Compound-Carryover in Shared Facilities" Pharmaceutical Online December 2017Walsh, A., “Cleaning Validation for the 21st Century: Acceptance Limits for Active Pharmaceutical Ingredients (APIs): Part I,” Pharmaceutical Engineering, Vol 31, No. 4, July/August 2011, pp. 74–83, available from: https://ispe.org/pharmaceutical-engineeringmagazine.
- Walsh, A., “Cleaning Validation for the 21st Century: Acceptance Limits for Active Pharmaceutical Ingredients (APIs): Part II,” Pharmaceutical Engineering, Vol 31, No. 5, September/October 2011, pp. 44–49,
- Walsh, Andrew, Ester Lovsin Barle, David G. Dolan, Andreas Flueckiger, Igor Gorsky, Robert Kowal, Mohammad Ovais, Osamu Shirokizawa, and Kelly Waldron. "A Process Capability-Derived Scale For Assessing Product Cross-Contamination Risk In Shared Facilities" Pharmaceutical Online, August 2017
- Docherty, S. E., “Establishing Microbial Cleaning Limits for Non-Sterile Manufacturing Equipment,” Pharmaceutical Engineering, Vol 29, No. 3, May/June 1999, pp. 36–40
- Walsh, A., “Microbial Aspects in Cleaning Validation,” in Microbiology and Sterility Assurance in Pharmaceuticals and Medical Devices, Madhu Raju Saghee, Tim Sandle, and Edward C. Tidswell, eds., Business Horizons, 2011, ISBN: 978-8-190646-74-1.
- Andrew Walsh, Thomas Altmann, Ralph Basile, Joel Bercu, Ph.D., Alfredo Canhoto, Ph.D., David G Dolan, Ph.D., Andreas Flueckiger M.D., Igor Gorsky, Jessica Graham, Ph.D., Ester Lovsin Barle,Ph.D., Ovais Mohammad, Mariann Neverovitch, Siegfried Schmitt, Ph.D., and Osamu Shirokizawa " The Shirokizawa Matrix: Determining The Level Of Effort, Formality, & Documentation In Cleaning Validation" Pharmaceutical Online, December 2019
- Walsh, Andrew, Thomas Altmann, Alfredo Canhoto, Ester Lovsin Barle, David G. Dolan, Andreas Flueckiger, Igor Gorsky, Jessica Graham, Ph.D., Robert Kowal, Mariann Neverovitch, Mohammad Ovais, Osamu Shirokizawa and Kelly Waldron "Measuring Risk in Cleaning: Cleaning FMEA and the Cleaning Risk Dashboard" Pharmaceutical Online, April 2018
- https://www.researchgate.net/publication/324259996_Spreadsheet_to_
Create_a_Toxicity_Scale_from_HBELs - https://www.researchgate.net/publication/324260079_Spreadsheet_to_
Create_a_Process_Capability_Scale_from_Cpu_Data - https://www.researchgate.net/publication/324260010_Spreadsheet_for_
create_Detectability_Scales_from_TOC_and_Visual_Inspection_Detection_Limits