
Increase In Drug Approvals, Priority Reviews Points To Bright Future For CMO Industry
By Adam Bradbury, industry analyst, PharmSource, part of GlobalData

New drug approvals are on the way up, as the FDA approved 11 percent more innovator therapies in 2018 compared to the previous year, spelling good news for both marketing authorization holders (MAHs) and CMOs. Data also shows that small- and mid-cap pharma companies are increasingly turning to outsourcers to manufacture newly approved drugs.
In 2018, the FDA approved 137 NDAs and BLAs, including new molecular entities (NMEs) and new formulations of older drugs, according to the GlobalData Drugs database. This figure represents an 11 percent increase over 2017 (122).
Overall, 57 NDAs were manufactured by contractors in 2018. In 2018, 51 percent of NMEs were outsourced, compared to 33 compared of non-NME NDA products.
The share of solid dose NMEs outsourced in 2018 stands at 52 percent, similar to the 2012-2017 average. However, outsourced injectable approvals increased slightly to 44 percent.
There were 22 dose CMOs that garnered contracts for NME products in 2018, with Catalent and Patheon (part of Thermo Fisher Scientific) topping the list with five and four contracts, respectively.
Mega-cap bio/pharma companies received 17 approvals in 2018 (12 percent of all NDAs). The overall number of approved mega-cap-sponsored NMEs remained fairly stable over the last decade, as did mega-cap companies’ propensity to outsource the manufacture of these drugs. NME outsourcing by small- and mid-cap companies markedly increased in 2018 compared with the 2013-2017 approvals average.
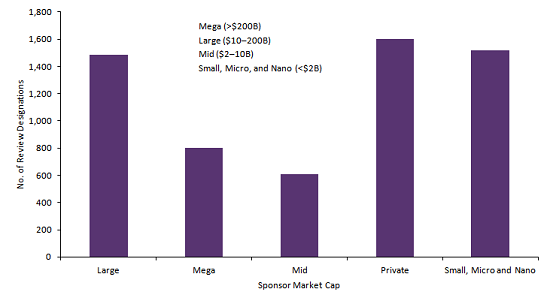
Figure 1: Outsourcing propensity for small-cap pharma companies, 2009-2018 (Source: GlobalData Drugs by Manufacturer [Accessed July 19, 2019] © GlobalData)
As shown in the figure above, small-cap pharma companies outsourced dose manufacture for 74 percent of NMEs and 70 percent of non-NME NDAs, which highlights the high level of dependence of small pharma on dose CMOs. The rate of outsourcing for the more innovative NMEs is slightly higher than non-NME NDAs, which shows that small-cap companies are less likely to have the expertise and/or technology to cope with manufacturing more innovative products. Out of the top four dose CMOs, Catalent was most dependent on private, mid-, and small-cap companies, whereas Patheon, Baxter, and Vetter manufactured most of their novel product approvals for large- and mega-cap companies in the past decade.
Priority Review And Other Accelerated Designations
NDA approvals were particularly high in 2018 for small-cap pharma companies, which sponsored 43 percent of NDAs, according to PharmSource research.1 As small biotech companies with limited budgets begin to dominate the industry, the FDA’s special designations, especially priority review, can provide valuable revenue streams of up to $350 million in cash to small and midsize biopharma firms. A priority review designation means the FDA’s goal is to take action on an application within six months (compared to 10 months under standard review). A priority review designation will direct overall attention and resources to the evaluation of applications for drugs that, if approved, would be significant improvements in the safety or effectiveness of the treatment, diagnosis, or prevention of serious conditions when compared to standard applications.
The FDA is awarding increasing numbers of priority review vouchers, according to GlobalData’s Regulatory Milestones Tracker database, which shows the number of priority reviews has risen by over 900 percent since 2012. Although this has decreased the sales price of priority review vouchers to an average of $80 million in recent years, in the context of small biotech companies, $80 million is no “small change,” and priority review vouchers continue to be a very useful fundraising tool.
For these cash-strapped companies, the revenue from these designations may be even more useful than the designations’ traditional benefits of extra regulatory help: regulatory-based incentives such as faster review and increased communication with the FDA are only useful if the company can afford to develop a drug.
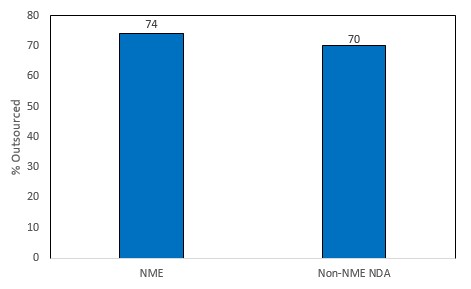
Figure 2: Number of FDA review designations awarded between 1985 and H1 2019, by sponsor market cap (Source: GlobalData Regulatory Milestones Tracker database [Accessed July 5, 2019] © GlobalData)
In 2018, 34 orphan-designated drug NMEs were approved, the highest between 2009 and 2018; this represented 53 percent of all NME approvals. Of these approvals, 65 percent were outsourced, the strongest outsourcing propensity for orphan NMEs since 2014. Mega cap companies sponsored five orphan NMEs, and only one of these had commercial dose outsourced. However, this drug (Pifeltro) was dual sourced and therefore was also being manufactured in-house. Orphan disease development is one of the most lucrative therapeutics areas, with accelerated/flexible development timelines, additional exclusivity, tax breaks, and the possibility of premium pricing due to low competition.
There were 23 NMEs approved with fast track status in 2018, significantly more than the 18 in 2017. Of these products, 16 (70 percent) were outsourced, which is an increase over the five-year average (2013-2017) of 58 percent. Between 2016 and 2018, there has been an increase in the number of fast track drugs approved. The number of NMEs approved with breakthrough therapy designation (BTD) in 2018 declined from 2017, with only 14 approvals recorded for 2018. However, outsourcing percentages of BTDs have never been so high since the program began in 2013 to expedite clinical development of drugs that demonstrate significant improvement against marketed therapies for the treatment of a serious or life-threatening condition.
There was considerable overlap between orphan, fast track, and breakthrough designations, with numerous NMEs approved in 2018 receiving all three designations. The number of pipeline drugs being assigned these review designations overall has been increasing over the last decade. While these designations do not guarantee regulatory approval and will form part of the clinical trial attrition rate, this can be viewed as a further positive sign that the market approvals with these designations will continue to increase.
Containment Substance Manufacture
Around 60 percent of high-potency APIs (HPAPIs) developed are for oncology; containment facilities are in high demand and will be increasingly so in the future as the oncology drug development pipeline continues to grow. NME approvals that required manufacturing containment rose significantly from 2017 to 27 products in 2018, of which 32 percent were outsourced. The category was driven by the approval of no less than eight protein kinase inhibitors and six cytotoxic drugs during the year. In general, the number of high-potency drug approvals has increased over the last decade, which may be a positive sign for CMOs, as small- and mid-cap companies lack the expertise for regulatory compliance and high-containment facilities and seek to outsource NMEs requiring special handling. CDMOs that provide contract HPAPI manufacturing services must be prepared to adopt, improve, and implement new protocols, equipment, training, and technologies to meet increasingly stringent risk reduction and regulatory compliance in HPAPI manufacturing.
Large CMOs have made significant recent investments in high-potency manufacturing, including Ajinomoto Bio-Pharma Services,2 Hovione,3 Lonza,2 and PCI Pharma Services.4 Controlled-substance manufacturing has seen similar interest, with investments by PCI Pharma Services;2 Noramco,5 which has also invested recently in controlled-substance manufacturing;2 Johnson Matthey;6 and Catalent.7
Catalent also completed the second phase of a $5.5 million expansion at its Kansas City, Missouri, clinical supplies facility. The CDMO has increased its controlled-substance and controlled-temperature storage capacity at the site. The first phase boosted its highly potent, cytotoxic, and cold storage clinical packaging capabilities; this latest expansion, completed in May 2018, includes a 3,600 square foot Drug Enforcement Administration (DEA) Schedule I and II controlled-substance vault, 450 controlled-temperature pallet locations, 500 high-density storage locations, and a new sampling room.
Priority review therapies and other accelerated drugs are more likely to be contract manufactured than products that go through standard review. Historically, dose manufacture for 70 percent of accelerated drugs has been outsourced versus an average of 45 percent for all drugs. Both small- and mid-cap companies are more likely to outsource their dose manufacture than large- and mega-cap companies due to a lack of in-house manufacturing capabilities and/or expertise. Therefore, the rising number of priority review designations and small-cap companies gaining FDA approvals can only be a positive sign for dose CMOs. High-containment substance equipment and facilities are prohibitively expensive and require a high level of expertise, and with increasingly stringent regulations as well, these forms of manufacture are also more likely to be outsourced. Overall, there are great opportunities for innovative CMOs that are able to adapt to manufacture increasingly complex drugs and produce drugs within shorter timelines required by certain increasingly used regulatory designations.
This article has been adapted from the 2019 CPhI Annual Report.
References:
- PharmSource, CMO Scorecard: Outsourcing of NDA Approvals and CMO Performance – 2019 Edition (2019), GDPS0025MAR
- PharmSource, Bio/Pharmaceutical Outsourcing Report (B/POR), March 2019
- PharmSource, Bio/Pharmaceutical Outsourcing Report (B/POR), March 2018
- PharmSource, Bio/Pharmaceutical Outsourcing Report (B/POR), November 2017
- PharmSource, Bio/Pharmaceutical Outsourcing Report (B/POR), November 2018
- PharmSource, Bio/Pharmaceutical Outsourcing Report (B/POR), October 2018
- PharmSource, Bio/Pharmaceutical Outsourcing Report (B/POR), June 2018
About The Author:
 Adam Bradbury currently serves as associate analyst for the PharmSource database, where his primary responsibilities include maintenance, improvement, and analysis of the contract manufacturing datasets. Bradbury has published numerous market research reports and has been involved in database improvements and integration for GlobalData’s Pharma Intelligence Center. He holds an MSc in human molecular genetics from Imperial College London and is located in GlobalData’s London office.
Adam Bradbury currently serves as associate analyst for the PharmSource database, where his primary responsibilities include maintenance, improvement, and analysis of the contract manufacturing datasets. Bradbury has published numerous market research reports and has been involved in database improvements and integration for GlobalData’s Pharma Intelligence Center. He holds an MSc in human molecular genetics from Imperial College London and is located in GlobalData’s London office.