
FDA FY2018 Human Tissue And Cell Therapy Observations And Trends
By Barbara Unger, Unger Consulting Inc.
 A comprehensive GMP intelligence program includes monitoring of health authority enforcement actions, including FDA Form 483s, Establishment Inspection Reports, warning letters, recalls, import alerts, consent decree agreements, and EU reports of GMDP noncompliance. This article presents the most recent publication of GMP Human Tissue for Transplantation Form 483 observation data from the FDA. The regulations that govern this area, 21 CFR 1271, are the same regulations that govern segments of human cell therapy and gene therapy. We examine data from FY2018 and evaluate a total of four years’ worth of trends in GMP inspection enforcement in this area.
A comprehensive GMP intelligence program includes monitoring of health authority enforcement actions, including FDA Form 483s, Establishment Inspection Reports, warning letters, recalls, import alerts, consent decree agreements, and EU reports of GMDP noncompliance. This article presents the most recent publication of GMP Human Tissue for Transplantation Form 483 observation data from the FDA. The regulations that govern this area, 21 CFR 1271, are the same regulations that govern segments of human cell therapy and gene therapy. We examine data from FY2018 and evaluate a total of four years’ worth of trends in GMP inspection enforcement in this area.
The presentation of data herein differs from how it is presented on the FDA website, even though it uses the same raw data. For example, I combine all observation listings within a given section that cite 21 CFR 1271 into a single value. For example, §1271.47(a) and §1271.47(b) are consolidated into a single line item, §1271.47. Similar consolidation is done for each of the §1271 citations identified in Table 1. Consolidating the citations leads to slightly different conclusions. In my opinion, sometimes, slicing the observations so narrowly, as the FDA does, can dilute their impact.
The FDA’s data includes only Form 483s issued through its electronic system. Thus, the data does not represent the FDA's complete collection of inspection observations for the year, and conclusions should be tempered by this limitation.
Also, for the collection of products covered under Human Tissue Transplantation, observations may also cite §211, particularly for cell and therapy products that are not “minimally manipulated” or are not destined for “homologous use”. Many of the firms that are cited for observations under §1271 are fertility clinics and the ever-increasing number of mostly unlicensed stem cell therapy firms. Many stem cell firms do not produce minimally manipulated or homologous use cells, yet they continue to distribute them commercially and run afoul of the FDA, including several firms that received warning letters this year. The FDA has published detailed guidance on what constitutes minimally manipulated and homologous use. Further, the FDA announced it is exercising “enforcement discretion” for three years after the governing guidance was published in November 2017, and it is only taking action when the products pose a clear public health risk. It seems that few of the firms are submitting INDs anticipating that this grace period will be over in late 2020, likely because too much money can be made in this area. It will be worth watching what actions are taken against these firms at the end of the enforcement discretion period.
Executive Summary
- Human Tissue Transplantation firms received only a fraction of the Form 483s issued to drug firms, and the number of 483s in this area varied from year to year by a larger percentage. See the previous article on drug GMP 483 observations. Particularly in FY2018, the number of Form 483s increased approximately 60 percent between FY2017 and FY2018.
- FY2017 appears to be an anomaly when considering the four years covered in this analysis. The total number of Form 483s was substantially lower that year, and the number of observations based on Donor Eligibility were almost half of what they were the preceding two years, and almost one-third of what they were in FY2018.
- Observations in the area of Donor Eligibility clearly outpace the number of observations in the area of Current Good Tissue Practices.
- The number of observations in the area of Current Good Tissue Practices fell by more than half in FY2018 compared to both FY2016 and FY2017 and was significantly lower than in FY2015.
- In contrast, the number of observations addressing Donor Eligibility increased significantly in FY2018 over any of the three previous years.
FDA Form 483 Inspection Observations
Figure 1 shows the number of Form 483s that have been issued in this product category over the last four fiscal years. FY 2017 saw many fewer issued than in any of the other three years. Overall, eliminating FY2017, the number of 483s issued has gradually increased since FY2015, peaking in FY2018. I would expect the number to continue to increase in FY2019.
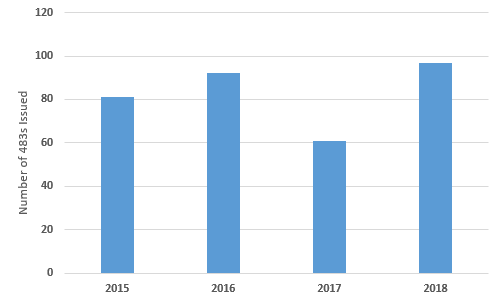
Figure 1: Total Form 483s issued between FY2015 and FY2018
Table 1 shows only the 11 most frequent inspection citations between FY2015 and FY 2018, while the tabulation on the FDA website shows all citations in the fiscal year. The FDA uses the term “frequency” to represent the number of times the agency identified a specific citation in its tabulation. Table 1 presents those observations for FY2018 modified as described in the Introduction section of this article.
Table 1: Human Tissue Product GMP Inspections, §1271 Citation Frequency
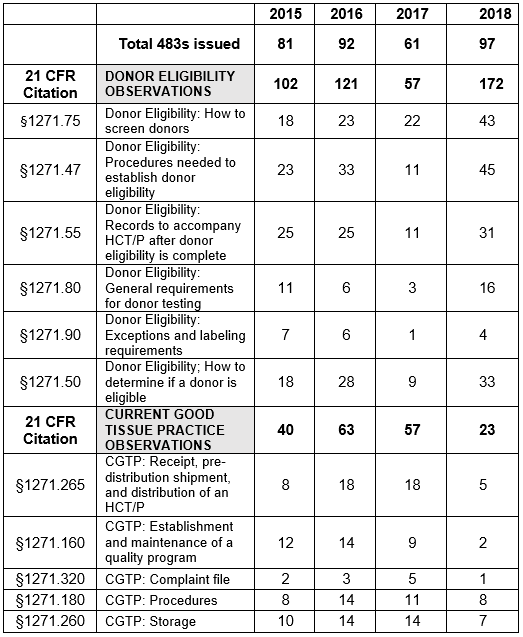
Figure 2 shows the total number of Form 483 observations in two areas, Subpart C Donor Eligibility and Subpart D Current Good Tissue Practices. From FY2015 through FY2017, the CGTP observations increased from 28 percent in 2015 to 34 percent in 2016 and 50 percent in 2017, then fell dramatically to 12 percent in 2018. Thus, most of the focus in FY2018 addressed failure to meet or adequately document donor eligibility requirements, including testing for infectious diseases or assessment for other risk factors. This may represent the FDA’s increasing focus on stem cell clinics, many of which operate outside of the regulatory environment during the period of inspection enforcement discretion.
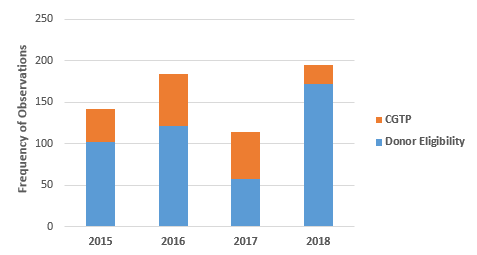
Figure 2: Donor eligibility vs. current good tissue practice observations
Figures 3 and 4 provide more granularity regarding the specific regulation areas cited for both Donor Eligibility and CGTP. Figure 3 provides the six most frequently cited sections for observations addressing Donor Eligibility. In all but one of the sections, FY2018 showed marked increases over the three previous fiscal years. The number of observations citing exceptions and labeling requirements, §1271.90, has decreased consistently from a high of seven in FY2015. As addressed above, the results from FY2017 seem to be an anomaly. The four most frequently cited sections in FY2018 have increased steadily over the years from FY2015 and include:
- §1271.47: Procedures needed to establish donor eligibility
- §1271.50: How to determine if a donor is eligible
- §1271.55: Records to accompany HCT/P after donor eligibility is completed
- §1271.75: How to screen donors
The two least frequently cited sections include §1271.80, general testing for donor requirements, and §12.71.90, exceptions and labeling requirements.
Figure 4 shows marked decreases in the five most frequent citations for FY2018 when compared to the three previous fiscal years. In all cases, the numbers have decreased from those in the previous year. Observations associated with the regulation to establish and maintain a quality program decreased from a high of 14 in FY2016 to two in FY2018.
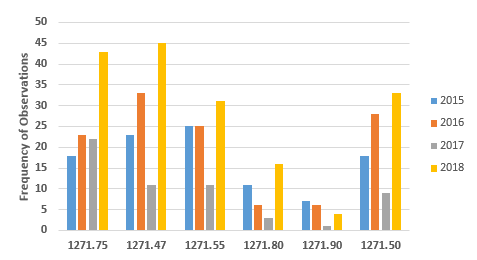
Figure 3: Observations associated with donor eligibility
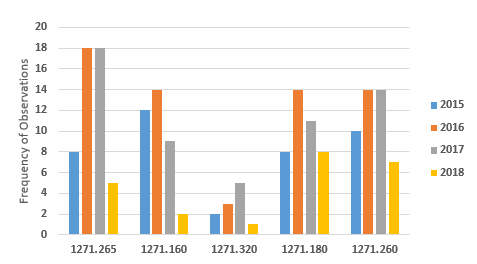
Figure 4: Observations associated with current good tissue practice
Conclusions
Human Tissue Transplantation will become a more important area for GMP inspections, reflecting the increase in the number of gene and cell therapy firms, including stem cell clinics, and is clearly worth watching over the coming years. When enforcement discretion for stem cell clinics ceases in late 2020, we may see a surge in these inspections and subsequent enforcement actions. This area can be complicated and includes the intersection of §1271 and §211, the GMPs for drug products, depending on the nature of the cell- or tissue-based product. The number of inspections, with the exception of FY2017, has increased over the past four years. The inspection focus for this group of products is primarily in the area of donor eligibility and the failure to document or perform adequate testing and medical record review of the donor. The potential infectious disease risks posed by human tissue or cell donations are responsible for this focus. CGTP observation frequency comes in a distant second to those in the area of donor eligibility. It will be useful to see if this trend continues in the coming years.
About The Author:
 Barbara Unger formed Unger Consulting, Inc. to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including general GMP auditing and auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She is currently the co-lead of the Rx-360 Data Integrity Working Group. You can contact her at bwunger123@gmail.com.
Barbara Unger formed Unger Consulting, Inc. to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including general GMP auditing and auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She is currently the co-lead of the Rx-360 Data Integrity Working Group. You can contact her at bwunger123@gmail.com.