
Do Patient Needs Define Biologic Product Specifications?
By Gerald Gellermann, André Mischo, Rainer Koeck, and Stephan Kirsch, Novartis Technical Research and Development

Understanding pharmaceutical products is crucial for ensuring patient safety and efficacy. Generating product knowledge and specifications to control critical variants or impurities is a lengthy, iterative process.
Early in technical development, after candidate selection, there is limited product knowledge, leading to high uncertainty about safety and efficacy. This uncertainty decreases as data is generated during technical and clinical development. When setting specifications for biological products, there has been a strong emphasis on batches used in preclinical and clinical studies to ensure that “the quality of the material made at commercial scale should be representative of the lots used in preclinical and clinical studies” (ICH Q6B)1.
This “conventional” approach was influenced by uncertainties due to the complexity of biologics products and limitations in generating detailed insights into all potential product variants and degradation products and their clinical relevance and safety.
While this conventional approach to defining product quality was in the best interest of patients, various challenges and limitations became apparent:
- It sets a standard expectation of analytical tests to be specified and does not reward the generation of enhanced knowledge to separate the critical attributes from non-critical attributes to address remaining uncertainties.
- Long product life cycles require manufacturing changes (e.g., raw materials, process scale-ups/transfers) that are essential to ensure product supply but can impact product quality beyond clinically validated ranges. Even if these changes have no clinically relevant impact, regulatory hurdles related to conventional, established specifications for analytical tests are often too high to implement, leading to shortages.
- Limitations on implementing process changes have also slowed down ESG-related improvements and innovations among pharmaceutical manufacturers.
- Specifications are closely connected to the analytical method used for clinical batches, making method-related changes very challenging to implement, even if required due to discontinuation of supplies (while essential for release and supply of the pharmaceutical product).
- Different approaches to determining specifications from clinical product quality often leads to registered specifications that vary by country, limiting the comparability of pharmacovigilance monitoring across countries.
Since the approval of ICH Q6B over 25 years ago, technological advancements have enabled more detailed molecular and structural characterization of biologics molecules and related product variants. Additionally, significant knowledge has been generated, especially for IgG-based mAbs and mAb-like formats.
These molecular insights, in addition to clinical validation, reduce uncertainties in complex therapies and manufacturing processes (see figure 1) by elucidating the structure-function relationship to distinguish critical quality attributes (CQAs) from non-critical ones. Technologies like AlphaFold, high-resolution X-ray diffraction, and deuterium exchange experiments support this elucidation. These advancements enable tailored control strategies for complex biologics products. However, the acceptance of these technologies for specification justification still needs to be integrated into the pharmaceutical regulatory environment.
Therefore, the enhanced approach to pharmaceutical quality must include transparent communication on how quality is continuously managed and ensured in the commercial phase, especially when product specifications are wider than manufacturing capabilities and justified based on risk-based impact assessments.
Figure 1: Different ways to ensure pharmaceutical quality
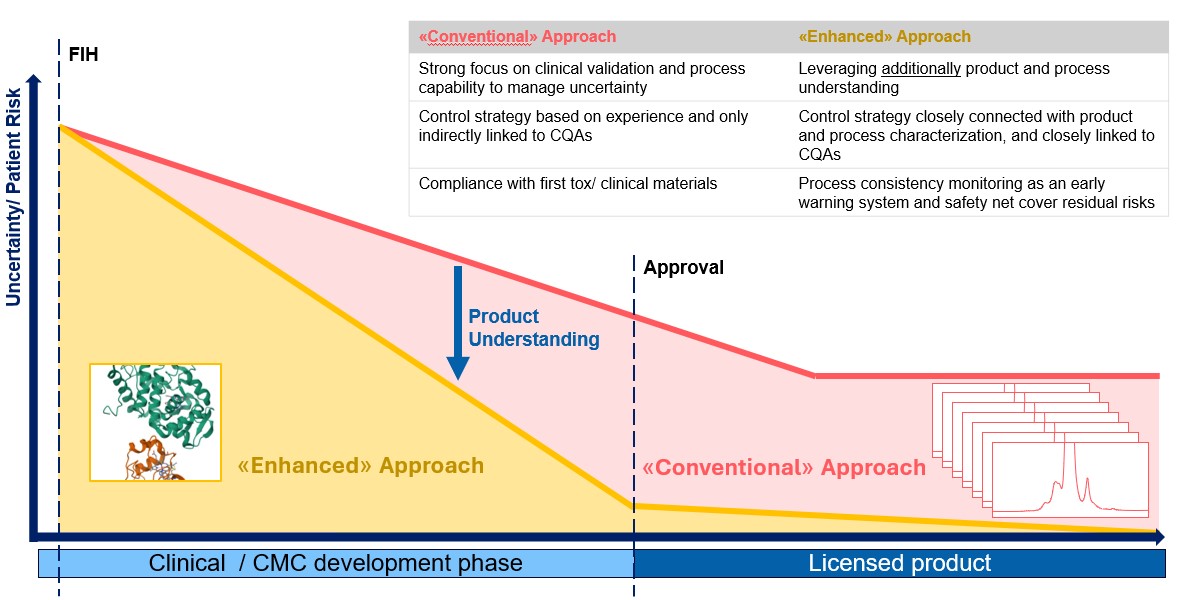
At the start of the clinical trial phase, safety uncertainty is at its highest. Several measures ensure an acceptable uncertainty level before trials begin. As trials progress, uncertainty about safety and efficacy decreases. At the CMC level, in-depth characterization and definition of critical quality attributes (CQAs) can reduce uncertainty more quickly and reliably. This allows for a more focused control strategy, enabling high control and less uncertainty in the event of a deviation or necessary change in the manufacturing process or analytical control.
In the enhanced approach, the conceptual distinction between product-specific and patient-centric specifications versus manufacturing data-based specifications for ensuring process consistency is becoming clearer. This distinction is also a motivation behind the ongoing revision of ICH Q6B.1 Different terminologies are often used for the same concept of a threshold that considers acceptable levels of CQAs, ensuring no difference in clinical outcomes for patients. Regardless of the terminology, in the enhanced approach, a CQA threshold is defined by solid product understanding informed by product characterization, stress studies, clinical batch analysis, and monitoring, as well as prior knowledge. As such, the threshold is defined to limit the impact of the CQA and can be analytical procedure-independent.
Process consistency control, on the other hand, does not necessarily consider product understanding. The optimal risk-based approach to pharmaceutical quality should use both elements, the product understanding-based specification definition and the process consistency control. Disclosing how both elements work together enables regulators to fully understand quality control over a product's life cycle and allows for the departure from tight specification limits for non-critical parameters. This, hopefully, enables manufacturers to overcome the above-mentioned limitations and ultimately benefits patients.
Conventional Approach To Specification Setting: ‘Purity’ Control For Biologics
Biological products like IgGs consist of many variants. Sensitive analytical technologies, such as ion exchange liquid chromatography, reliably quantify different charge variant groups (figure 2). This technology detects differences in charge distribution from manufacturing variability or product degradation during storage. Consequently, charge assays monitor process consistency with respect to historical charge profiles. Defining quantitative batch acceptance criteria for major charge variants, such as sum of acidic variants, sum of basic variants, and main variants, allows for limiting the content of variant groups in future batches to those present in clinical trial material. As a result, authorities often request tight batch reject limits for the peak groups.
Figure 2: Charge separation assay (CEX) of an IgG
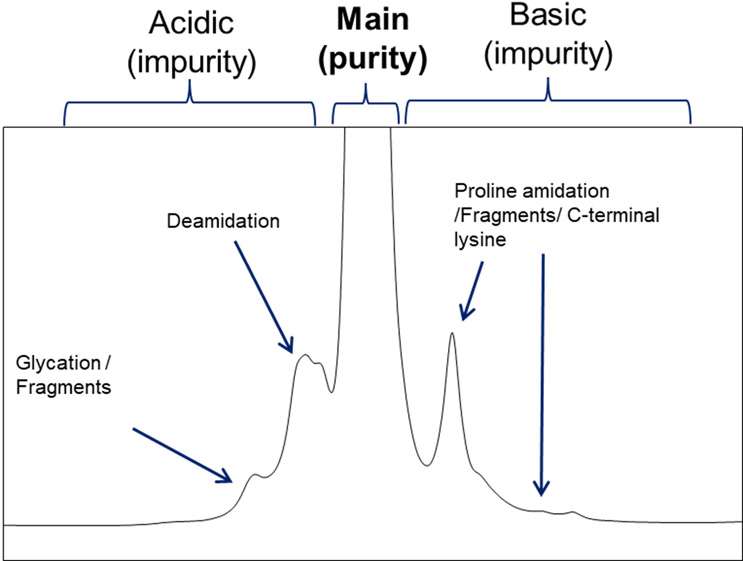
Typical charge assay chromatogram of a temperature-stressed IgG molecule. CEX separates variants based on charge distribution, enabling grouping them into acidic, basic, and main variants.
In the conventional approach, the biggest peak group in the chromatogram is referred to as “purity” (desired species), while the other two peak groups are labeled as impurities (undesired species). However, this concept does not consider the product understanding achieved through thorough product characterization and assessment of critical variants.
In biologics, many separated variants in the chromatogram are uncritical, while critical variants are often present in all three peak groups (including “purity”). Moreover, some IgGs have no “main variant,” and all three peak groups can be present in equal quantities. Different variants within the same peak group may even have opposite impacts on biological activity. Only peak characterization and testing in different functional assays increase product understanding and enable a tailored control system for pharmaceutical quality.
Product Characterization As A Basis For An Enhanced Approach To Define Specifications
To elucidate degradation pathways of the IgG in the example, the IgG was exposed to different stress conditions such as pH, temperature, freeze/thaw, light, and shaking. These stress conditions cover and exceed potential exposures the molecule might encounter during manufacturing and distribution until it is administered to a patient. Peak fractionation from the CEX (figure 2) combined with peptide-mapping mass spectrometry and functional potency assays helped to detect residues crucial for maintaining the desired biological function and evaluate their susceptibility to modification. The results and conclusions were supported by point mutations conducted during the research phase, as well as crystallization experiments of the IgG-Fab bound to its target. These experiments resulted in a high-resolution model of the structure that was further used to evaluate binding kinetics through in-silico modeling. This information was used conservatively to determine the CQAs. The following table provides a high-level overview of the species labeled in the chromatogram shown in figure 2.
Table 1: Peak characterization and CQA assessment of the IgG
| Variant/peak | Product understanding based on characterization experiments |
| Glycation | Highly glycated samples behaved comparably to the non-glycated IgG1 in representative drug to target binding studies. As glycation also occurs on natural human IgGs, the variant was determined as not critical. |
| Deamidation | Deamidation under various stress conditions occurred mainly in residues that are not involved in target binding or effector functions. The related variants could be clearly associated in an increase of the labeled peak of the acidic variants. Deamidation also occurred in smaller quantities in the paratope region (CDR). This deamidation (deamidation in the CDR) is a CQA and is further discussed below. |
| Proline amidation/c-terminal lysine | These residues do not influence target binding or effector functions and are thus not critical for this IgG. Resulting changes in charge are found to be lower than what is described as risk for PK.2 |
| Fragmentation | Fragments show changed bioactivity and PK properties in experimental in vitro studies and in accordance with general literature. Required analytical performance to control fragments is achieved by other technologies such as CE-SDS that better separate the fragments that occur in accordance with the characterized degradation pathways of the IgG. |
Based on the acquired product understanding, it can be concluded that CEX in this example is a suitable analytical procedure in the control system to monitor general process consistency regarding charge variant distribution but has only limited relevance for directly controlling CQAs such as fragments or deamidation in CDR. Fragments of the example IgG are controlled by other analytical methods that allow a direct quantitation, and deamidation in CDR does not require confirmatory routine analytical batch release control testing as described in the following section.

Considering that the IgG has two binding regions (two Fab arms with identical CDRs) but only one of them was found to be required for the determined IgG bind and block mechanism, an increase of deamidation up to 10% results in a potency impact of only 1% (based on a binominal distribution, at a 10% measured level, only 1% of the molecules are modified in both arms). Conservatively considering potential avidity effects, an acceptance criterion of 5% for deamidation in CDR is defined. As deamidation is a naturally occurring variant of human IgGs and a common modification in several therapeutic proteins, the 5% limit poses a low risk for altered immunogenicity and safety. This assessment was further supported by in-silico immunogenicity evaluations and clinical results showing the variant at levels from 1% to 2.4%. In addition, since the evaluated deamidation occurs in the CDR and not in areas relevant for the pharmacokinetic (PK) properties, the risk of altered PK at a 5% level is considered low. Hence, it is concluded that from a patient perspective, there are no differences in clinical outcomes if a level of 5% is not exceeded. This assessment is in line with similar approaches described in literature.3-8
Patient-Centric Control System To Ensure Pharmaceutical Quality
Based on the knowledge obtained from studies supporting the CQA assessment, stability testing of long-term stored representative samples, and predictive stability estimates for worst-case scenarios9, the likelihood that the variant increases by more than 3% to levels above 5% during manufacturing, storage, transport, and administration was determined to be very low.
The combination of worst-case considerations allows for the definition of a robust control strategy for deamidation in CDR, ensuring that inherent process and stability variability will not exceed the critical patient-relevant limit. Based on that, it is concluded that under routine manufacturing conditions, no confirmatory testing specifically designed for deamidation in CDR is required, as the risk of exceeding the 5% threshold is inherently low for routine manufacturing.
Nevertheless, manufacturing processes for biologics are complex and consist of multiple manufacturing steps. Process characterization and validation allow for the definition of process parameter acceptance criteria to control the inherent variability. However, unpredictable events such as operator failure or changes in environmental conditions can occur. To address this remaining unpredictable uncertainty, CEX testing is employed in the control system.
As outlined in the example, the CEX assay is generally sensitive to various changes, including predominantly uncritical residues that react more sensitively than the critical deamidation in CDR. Therefore, testing CEX at relevant points in the manufacturing chain is suitable for monitoring general process consistency and detecting potential unpredictable events that could impact the charge pattern. Monitoring limits, managed in the pharmaceutical quality system (PQS), build on historical manufacturing values and statistical assessments. Any deviation from the monitoring limit or statistical probabilities needs to be investigated to ensure that the predetermined low risk of the deamidation in CDR exceeding the patient-centric threshold remains low. The same applies to necessary changes, where an effectiveness check needs to be conducted to confirm that the risk profile for deamidation in CDR remains unchanged. Monitoring limits to ensure process consistency in this control system should be managed within the PQS and are not registered as fixed boundaries. If required by an authority, the approach to how they are managed within the PQS can be provided, e.g., in the dossier or during an inspection. This provides more flexibility to adapt innovations and support changes that have no impact on CQAs.
It is acknowledged that in cases where enhanced product understanding cannot be established, the limit definition for a sensitive analytical technology should not be wider than what was tested during clinical trials. Those limits need to be fixed and agreed upon with health authorities to address the remaining uncertainty of variant control for complex biologics.
From a regulator's perspective, it is crucial to understand how both elements, process consistency and patient-centric specifications, are interconnected and utilized in the patient-centric control strategy. This understanding is essential to accept why a wider patient-centric limit (e.g., 5% for deamidation in the binding region) poses no risk to patients.
Conclusion
Enhanced product understanding, when integrated into a patient-centric control system, offers significant benefits to both patients and industry. Generating in-depth knowledge to address the inherent complexity of biologics products can further reduce uncertainty around the safety and efficacy of pharmaceutical products. Additionally, it can provide manufacturers with more flexibility, enabling faster necessary changes to innovate, decrease environmental footprint, and extend storage times for home use or new devices, which represents a significant benefit to patients. However, greater transparency on how product understanding and controls work together is required.
The authors hope this article stimulates the revision of ICH Q6B1, which can later provide more guidance to the industry on how both elements, control of critical quality attributes versus process consistency control by using established analytical procedures, can be used in a combined way.
References:
- ICH Q6 and revision (R1): Specifications, available on internet: ICH Official website: ICH
- Bumbaca, D., et al., Physiochemical and Biochemical Factors Influencing the Pharmacokinetics of Antibody Therapeutics. The AAPS Journal, 2012. 14(3): p. 554-558
- Mire-Sluis A, Dobbins J, Moore CMV, Pepper T, Rellahan B, Riker K, Roberts M, Schultz T. Patient-Centric Quality Standards. J Pharm Sci. 2024 Apr;113(4):837-855. doi: 10.1016/j.xphs.2024.01.006. Epub 2024 Jan 25. PMID: 38280722.
- Philip Krause, Cristiana Campa, Andrew Chang, Shawn Novick, Barbara Rellahan, Tim Schofield, Dean Smith, A vision for patient-centric specifications for biologicals, Biologicals, Volume 88, 2024, 101796, ISSN 1045-1056, https://doi.org/10.1016/j.biologicals.2024.101796.
- Kretsinger J, Frantz N, Hart SA, Kelley WP, Kitchen B, Novick S, Rellahan B, Stranges D, Stroop CJM, Yin P, Gastens MH. Expectations for Phase-Appropriate Drug Substance and Drug Product Specifications for Early-Stage Protein Therapeutics. J Pharm Sci. 2019 Apr;108(4):1442-1452. doi: 10.1016/j.xphs.2018.11.042. Epub 2018 Dec 6. PMID: 30528942.
- Kepert JF, Cromwell M, Engler N, Finkler C, Gellermann G, Gennaro L, Harris R, Iverson R, Kelley B, Krummen L, McKnight N, Motchnik P, Schnaible V, Taticek R. Establishing a control system using QbD principles. Biologicals. 2016 Sep;44(5):319-31. doi: 10.1016/j.biologicals.2016.06.003. Epub 2016 Jul 16. PMID: 27430904.
- Gellermann, G; Rösli, C. Critical Quality Attribute Assessment and Definition of Acceptable Limits for IgG1 Monoclonal Antibodies. In Quality by Design—An Indispensable Approach to Accelerate Biopharmaceutical Product Development, Khan, M A; Campa, C, Eds. Parenteral Drug Association, Inc.: Bethesda, Md., 2021; pp 33-49.
- Riley, C. M., Nguyen, K. L., Gellermann, G., & Kirsch, S. (2025). Introduction. In Specification of Drug Substances and Products (pp. 3-18). Elsevier
- Kuzman D, Bunc M, Ravnik M, Reiter F, Žagar L, Bončina M. Long-term stability predictions of therapeutic monoclonal antibodies in solution using Arrhenius-based kinetics. Sci Rep. 2021 Oct 15;11(1):20534. doi: 10.1038/s41598-021-99875-9. PMID: 34654882; PMCID: PMC8519954.
 About The Authors:
About The Authors:
Gerald Gellermann works as a scientific officer at Novartis Analytical Development and, among other activities, leads initiatives related to the implementation of new ICH guidelines, such as ICH Q12 and Q14. Gerald holds a Ph.D. in molecular biology from the University of Jena, Germany.
 André Mischo works in the scientific office at Novartis Analytical Development. He has a strong interest in setting specifications and advocates the use of product understanding for this purpose. He holds a Ph.D. in biochemistry from the University of Jena, Germany.
André Mischo works in the scientific office at Novartis Analytical Development. He has a strong interest in setting specifications and advocates the use of product understanding for this purpose. He holds a Ph.D. in biochemistry from the University of Jena, Germany.
 Rainer Koeck works as a portfolio head at Novartis RA CMC Biologics and has a background in analytical development and the establishment of control systems. He is actively involved in supporting the ICH Q6 revision efforts and holds a Ph.D. in analytical chemistry from the University of Innsbruck, Austria.
Rainer Koeck works as a portfolio head at Novartis RA CMC Biologics and has a background in analytical development and the establishment of control systems. He is actively involved in supporting the ICH Q6 revision efforts and holds a Ph.D. in analytical chemistry from the University of Innsbruck, Austria.
 Stephan Kirsch leads the scientific office at Novartis Analytical Development and has a strong focus in quality by design, CMC development, and analytical technologies. He holds a Ph.D. in biochemistry from the University of Münster, Germany.
Stephan Kirsch leads the scientific office at Novartis Analytical Development and has a strong focus in quality by design, CMC development, and analytical technologies. He holds a Ph.D. in biochemistry from the University of Münster, Germany.