
De-Risking Your Bioconjugate's Development Path From Discovery To The Clinic
By Raphael Frey, Team Lead, Early Development Bioconjugates; and Sandro Holzer, Head of Process Development, Bioconjugates, Lonza Pharma & Biotech

Bioconjugation, in its broadest definition, is the chemical modification of biomolecules to enhance or alter the properties of a biomolecule. The biomolecule can be any biological entity, and the modification typically occurs via a reactive handle that sits on the biomolecule and selectively reacts with a linker payload molecule to form a covalent bond. A prominent type of bioconjugate is antibody-drug conjugates (ADCs), which is a powerful class of therapeutic agents in oncology and hematology that is gaining momentum in today’s market. Seven ADCs have been approved just since 2019, and recent investment activity between key players in the industry signal an emerging area with numerous opportunities.
Nevertheless, despite the promise of these biopharmaceutical Trojan horses, the drug development road for a bioconjugate is an arduous one, with many potential challenges and pitfalls throughout all of the life cycle stages, from lead discovery to market approval. Understanding the obstacles you may face in your journey as well as the tools available to help overcome them can help streamline your product’s path to market, facilitating the growth and distribution of this expanding class of innovative therapies.
ADC Development: Lessons From Past And Future Trends
In a new era of biomanufacturing, advances in science and technology are creating a diverse and complex landscape of novel therapeutics that offer renewed possibilities ― and hope― to patients across the globe. Making up approximately 10% to 15% of the biologics market, bioconjugates have an important role in this evolution.1 This includes not only the well-known ADCs but also a range of other bioconjugate modalities, such as PEGylated, radiolabeled, and polysaccharide conjugates. However, the manufacturing and development of bioconjugates is complex, calling on a range of different expertise and capabilities.
Focusing specifically on the emerging class of ADCs, Figure 1 outlines the factors that can impact the safety, efficacy, and manufacturability of each component that comprises these novel drug products. The added complexity of developing and manufacturing an ADC combined with the current success rate of a drug candidate, from the beginning of the clinical trial to receiving marketing approval, at only about 10%-20%, makes overcoming the challenges of ADC development critical. It also underlines the importance of choosing robust and scalable technologies at the very onset of a drug development program.2
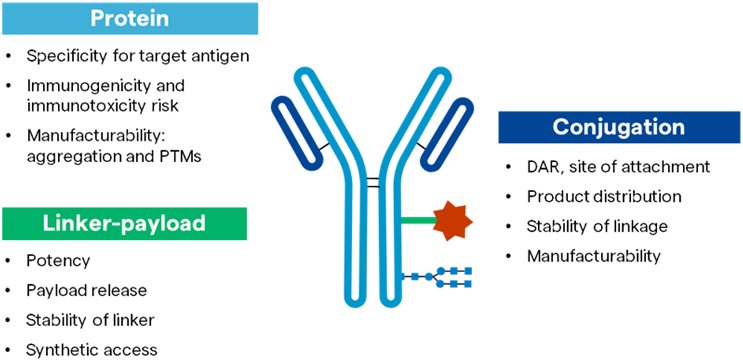
Figure 1: Efficacy, safety, and manufacturability of an ADC depends on each component.
Looking back at the history of ADC development, early-generation ADCs often showed poor performance in the clinic. This was due to several reasons, including low payload potency, linkers that were too labile, and conjugation processes that were not optimized enough.3
With added knowledge about ADCs and new technologies, highly cytotoxic drugs, such as auristatins and maytansine, became more accessible over time and were eventually developed as ADC payloads. New linker technology was also developed, offering stable linkers that are cleavable and allowed release upon internalization of the antibody. The advancements during this decade of development eventually led to the first approved ADCs on the market. Continued efforts since have yielded new technologies, including payloads, linkers, and conjugation methodologies. These developments, and more, will likely improve the therapeutic window of ADCs further.4
Bioconjugate Development Today
Today’s pipeline of approved ADCs (Figure 2) still reflects the state of technology as it was a decade ago, with molecules based on a limited set of technologies, such as stochastic conjugation to reduced interchain disulfide bonds. This typically leads to a distribution of products with a drug-to-antibody ratio (DAR) from 0 up to 8 (often aiming at an average DAR of around 4). There are also ADCs based on stochastic conjugation to lysine residues, which typically adds to the product heterogeneity.
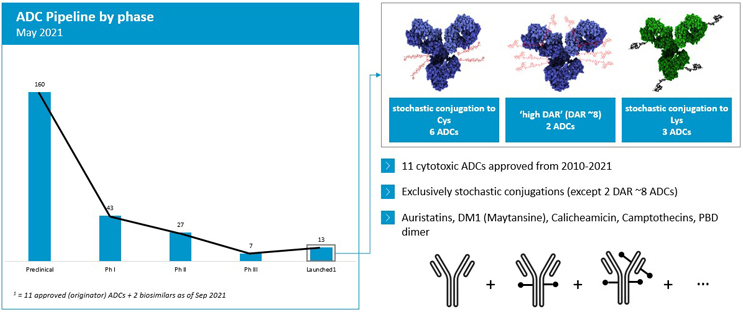
Figure 2: ADC pipeline breakdown by phase
In contrast, site-specific conjugation technologies yield more homogeneous products as modification with the payload-linker occurs at a defined site on the protein. ADCs conjugated site-specifically are often associated with improved pharmacokinetic properties and efficacy in vivo.5,6 Breaking down the proportion of ADCs entering the clinic by conjugation technology, as shown in Figure 3, provides insight into where the market is headed.7 Blue indicates the proportion of molecules based on site-specific technology, while gray shows those molecules based on non-site-specific technology, showing a steady increase of ADCs based on site-specific technology entering the clinic. This emphasizes the potential that many drug developers see in using site-specific technology to create better therapies.
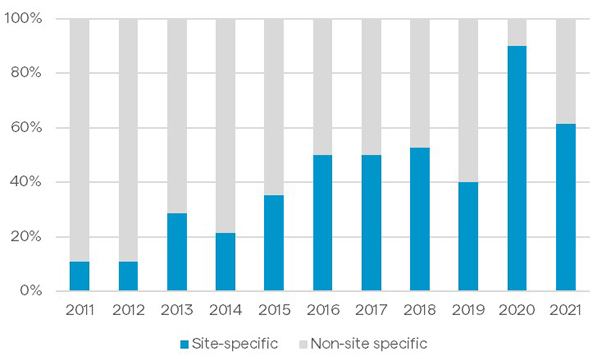
Figure 3: Proportion of ADCs entering the clinic by conjugation technology (ADCs with undisclosed conjugation approaches are excluded)7
It is important to note that while there is a diverse and growing range of technologies that has contributed to this trend (Figure 4), stochastic conjugation will remain a relevant and important method for years to come.
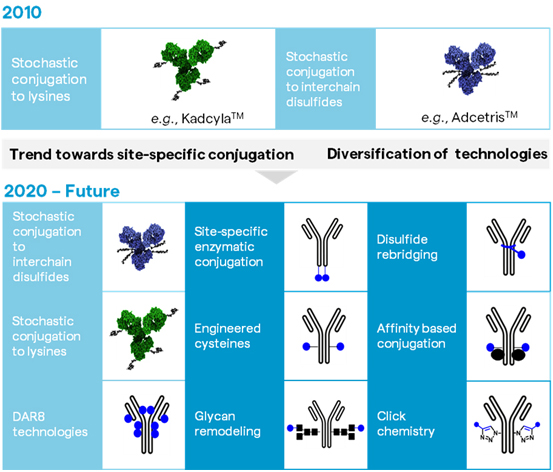
Figure 4: The evolution of conjugation technologies over the last decade. It is important to note that this summary is non-exhaustive, as only a few representative examples of site-specific conjugation methods are shown in the image.
In addition to the growing market for ADCs, there is a healthy pipeline of other modalities, including a range of non-toxic payloads that create opportunities to address other disease areas using bioconjugation. Alternative target-binding formats, such as smaller antibody-like proteins, also drive new possibilities for issues associated with the classical immunoglobulin scaffold. These developments emphasize the continued need for technologies that are not limited to a particular protein or specific linker payload but can be used for a range of different applications.
Navigating today's options of bioconjugation technologies is a challenge for companies entering this area of the market, which is why Lonza offers its clients a selected range of state-of-the-art technologies from an early stage, all proven to be scalable and applicable to different modalities. This includes development tools for the protein and linker as well as those needed for bioconjugation; a strong focus on scalability and robustness allows for a smoother journey in the drug development process, especially as scale-up becomes more important.
Building Bioconjugate Leads Using Lonza’s Development Toolbox
Lonza’s toolbox of bioconjugation technologies includes both conventional and site-specific conjugation. Lonza developed a proprietary platform of antibodies with engineered cysteine residues that allows for the production of specific homogeneous DAR 2 and DAR 4 products. In addition to Lonza’s proprietary platforms and expertise, which goes beyond classical ADCs, the team also collaborates closely with technology partners to make innovative conjugation technologies accessible to Lonza clients. As part of such a collaboration, Lonza runs an in-depth evaluation of the external technology in their development labs to assess suitability of the technology for lead generation as well as its robustness and scalability in order to mitigate risk in the later stages of development.
In regard to linkers and payloads, Lonza has a strong track record in the synthesis of cytotoxic drug linkers, with 15 ADC payloads and 50 highly potent APIs manufactured for both clinical and commercial products since 2009. For clients in the early stages of their project, Lonza has trusted suppliers to source commercially available cytotoxic molecules and linkers, thereby granting ready access to preclinical materials. This is often more resource- and time-efficient than a custom synthesis for the lead-generation and proof-of-concept phase.
Finally, Lonza’s protein experts in early development services located in Cambridge, UK, developed a range of in silico and in vitro tools to optimize and de-risk the protein for Lonza clients’ bioconjugates. With these tools, the team selects the candidate with an optimal specificity profile, evaluates and addresses immunogenic and immunotoxicity issues early, and completes a manufacturability assessment, including aggregation and post-translational modifications. Along with these design and protein optimization tools, Lonza’s protein expression services can facilitate access to non-GMP material for preclinical needs and, eventually, expression of GMP material.
An Integrated Solution For Early-Stage Development Of Bioconjugates
All of Lonza’s tools and capabilities outlined above can be combined to create an integrated solution for early-stage development of bioconjugates. This solution allows clients to move from an initial bioconjugate design to a lead candidate in two stages (Figure 5).
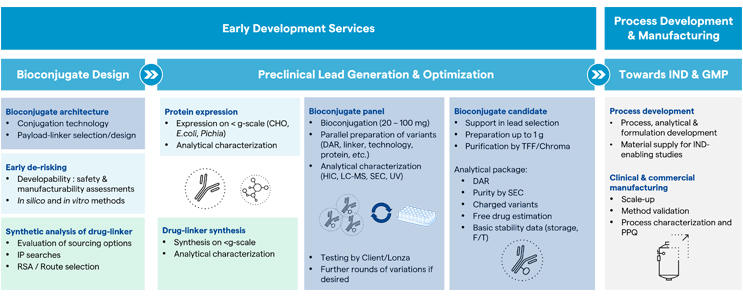
Figure 5: Lonza provides its clients with an integrated solution from initial bioconjugate design to a lead candidate in two stages.
The process begins with the bioconjugate design, which focuses on bioconjugate architecture, looking specifically at which technologies best suit the product requirements. This phase also includes the option for de-risking the protein using the developability assessments described above. Finally, a strategy is developed to gain access to the drug linker, either via synthetic routes or using commercial sourcing options.
The lead generation and optimization phases begin with the expression of the protein and, in parallel, the drug linker synthesis or sourcing. These components are then brought together in Lonza’s bioconjugation facilities where multiple milligram-scale reactions can be run in parallel to establish the conjugation process, test different technologies, and/or vary certain product attributes, such as the payload-to-protein ratio. The resulting panel of bioconjugates can be tested for efficacy in-house by Lonza or by the client. Those candidates with promising properties can undergo additional rounds of variations, if necessary, until an optimized lead candidate is obtained. In order to support further in vitro and in vivo testing, Lonza’s bioconjugation scientists can scale up the reaction and supply gram-scale amounts within approximately four weeks, including a basic set of analytical data. Once a drug candidate is identified, Lonza will identify the steps necessary to move a program through process development and toward the IND submission, maintaining a continued focus on scale-up and manufacturing.
Development Pathways For Bioconjugation
Even with the selection of a lead candidate, the bioconjugate development journey does not end. In the next stage, the main goal is to move from the process that was applied to obtain the selected bioconjugate, to the manufacturing process that allows for GMP production. The manufacturing process should fulfill certain key criteria, such as robustness, scalability, and reproducibility, which is achieved by using thorough process development strategies. Timelines to the IND submission and first clinical studies are usually tight, so a selected set of well-designed experiments is needed. Avoiding iteration and repetition of experiments is crucial.
Figure 6 is a schematic overview of the various development paths that can lead to the manufacture of clinical material.
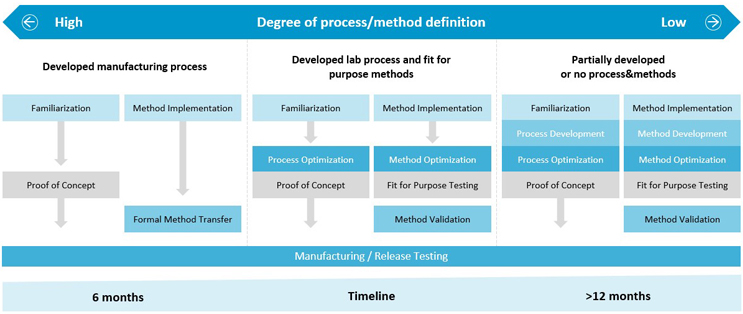
Figure 6: Lonza’s process development paths for bioconjugates
Lonza collaborates with its customers to provide a tailored development solution that ensures successful transfer into GMP assets and addresses any gaps based on the degree of process and method definition. The appropriate development path will determine the development timeline.
An integrated offering with early development services brings several advantages. First, fast internal handover of process and analytics is possible, with knowledge history on the process and analytics shared internally. In addition, the selected technologies and methods are well aligned with Lonza’s development and manufacturing capabilities. These synergies prevent lost time through additional interfaces, redundancy, and necessary re-optimizations.
Challenges For The Development Of A Bioconjugate
Many challenges are possible on the journey to a bioconjugate manufacturing process, such as the need to establish robust safety procedures when working with cytotoxic substances or an analytical toolbox that can appropriately monitor the critical quality attributes and enables GMP release of a drug substance. The following case studies detail how Lonza used its capabilities to control critical quality attributes of a highly cytotoxic ADC and achieve scalable purification techniques, while maintaining criteria for robustness, reproducibility, and scalability throughout the development process.
Case Study 1: Conjugation Of A Highly Toxic ADC
Figure 7 outlines the process of stoichiometric reduction and subsequent conjugation to free thiols as it was transferred from the customer to Lonza.

Figure 7: Transfer of customer process to Lonza for the identification of optimization targets
The customer had already performed several batches at small scale with the goal of GMP production to supply for clinical Phase 1. Lonza’s team assessed these process steps for scalability and manufacturability, identifying several parameters that presented risks for a scale-up.
Optimization Target A: Reaction Buffer
Buffers are used to assure a stable pH throughout the manufacturing process, as well as in the formulation. While antibodies, including ADCs, are usually most stable in a pH range from 5 to 6, the optimal process pH depends on the type of reaction. The chosen buffer must have a strong buffer capacity in that pH range. However, the reaction buffer used in the original process did not fulfill these criteria.
To find a more suitable buffer, several buffer compositions based on their chemical properties were explored. They contained either phosphate, histidine, or TRIS and also covered different pH ranges. Experiments were performed at a small scale in scale-down reactors, allowing the parallel screening of multiple conditions. Analytical data, such as the DAR profile and aggregation levels, were then evaluated.
When comparing the DAR profile of the initial reaction buffer to the optimized reaction buffer, two improvements were observed. First, the amount of uneven DAR species was initially elevated, which is unusual for cysteine conjugations. This indicated an incomplete conjugation or side reactions. With the new reaction buffer, the uneven DAR species were significantly reduced. The second observation was a shift toward high DAR species, showing an overall increase in conjugation efficiency.
Optimization Target B: Tangential Flow Filtration (TFF)
TFF is used in order to remove any drug-related impurities, to concentrate the product or to exchange the buffer. The process solution is circulated from a vessel to a filter membrane, where it moves tangentially to the membrane. Pore size is large enough to let impurities permeate through this membrane but small enough to retain the product. The key advantage of TFF is that the process step and its parameters are scalable. The parameters that must first be optimized during process development include trans membrane pressure (or TMP, which corresponds to the pressure drop through the membrane), feed flow, membrane loading, and protein concentration.
Lonza’s team conducted two experiments to optimize TFF for the customer. In the first experiment, screening was performed for a combination of TMP and feed flow rate that maximized filtrate flux (Figure 8). The second experiment was to optimize the number of diavolumes. A maximum number of diavolumes was performed, and free drug and solvent were measured at different pull points. The goal was to choose the minimum number of diavolumes needed for adequate clearance, which, in this case, was achieved after 10 diavolumes.
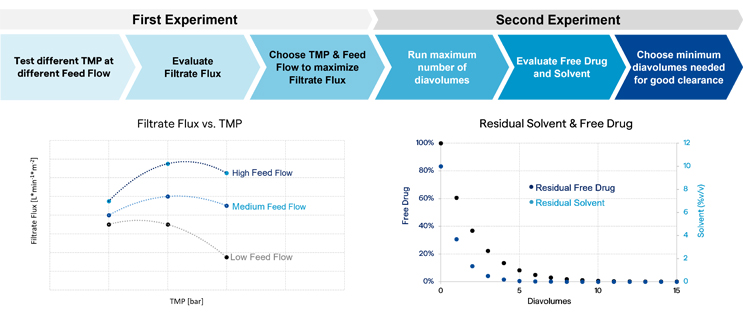
Figure 8: Optimization of process parameters for diafiltration
As a result of this optimization, stress on the product, process time, and buffer consumption can be reduced.
Case Study 2: Chromatography Development
Another powerful and scalable purification technique is chromatography. This method is used to effectively remove drug-related impurities, residual solvents, and aggregates. With more sophisticated purification protocols, an enrichment of specific DAR species is also possible. Consequently, chromatography can be used as a tool to improve process robustness by absorbing some of the resulting variability from the chemical process steps. In order to develop suitable chromatography purification, several factors need to be considered. These include resin selection, binding capacity, separation, pooling criteria, buffer consumption, and cycle time.
Figure 9 outlines Lonza’s process for developing or optimizing polishing chromatography after bioconjugation.
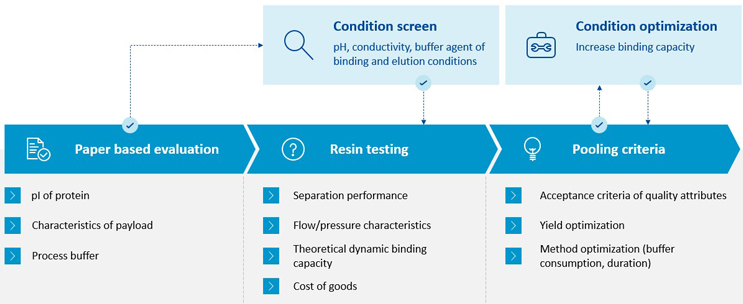
Figure 9: Chromatography development for bioconjugation processes at Lonza
When developing or optimizing polishing chromatography after bioconjugation, the first step is a paper-based evaluation of the separation problem assessing the characteristics of different components and narrowing resin types for selection. In situations of a simpler resin/product interaction, the next step would be to move directly to small-scale resin testing, where a subset of resins is evaluated based on separation performance, flow/pressure characteristics, theoretical dynamic binding capacity, and cost of goods. However, in more complex cases, where prediction of binding/elution behavior is more difficult (e.g., mixed mode), a condition robot screening is performed first. Once the resin and initial conditions are defined, the team performs chromatography runs on a representative bed height. This is to achieve comparable separation behavior as with the manufacturing run. Based on this data, the pooling criteria can be defined. Lastly, further optimization can be achieved by iterative condition screenings.
In this case study, a customer had developed a conjugation process but also wanted to have a step developed to polish the product. After completing the paper-based assessment mentioned above, the team selected two resins that were deemed suitable for the intended purpose. Using the pipetting robot, binding elution conditions on two different resins were screened. Figure 10 shows the various combinations of UV absorption throughout the different phases of the chromatography method.
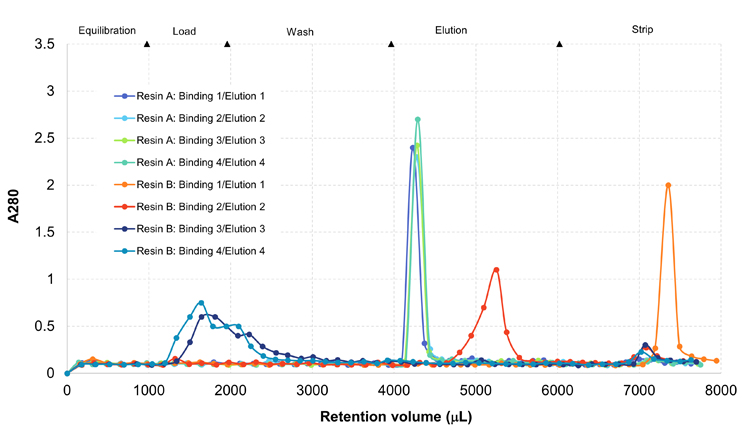
Figure 10: Resin and condition screening
Some combinations did not exhibit binding or struggled to elute the protein from the column while others (around 4000 µL retention) indicate protein binding and elution. Next, the selected condition resin combination was tested on small-scale columns. This is done not only to confirm the screening results, but also to gain insights on separation performance, i.e., information that cannot be gathered from the screening.
Separation data from small-scale columns is depicted in Figure 11. Applying a pH gradient did not lead to any separation of the impurity peaks (Fig 11, top left), whereas with a salt gradient, partial separation was observed (Fig 11, top right). As a consequence, a salt gradient was further investigated. Next, experiments continued on a bed height representative of manufacturing, allowing definition of pooling criteria and optimization of yield. Figure 11 also shows a fractionated elution peak from a column that reflected the situation in GMP (relevant quality attributes of the product analyzed in each fraction). Here, the high and low molecule weight species were tracked, and pooling criteria was selected that allowed appropriate control over aggregates in the product. This information also allows estimates on yield impact if a fraction is rejected.
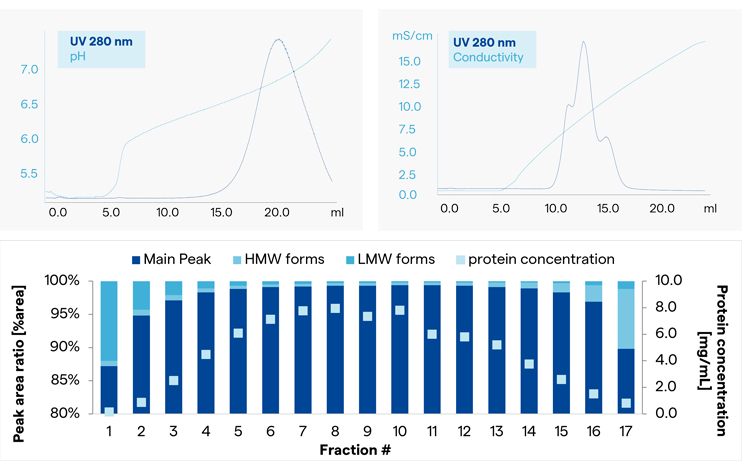
Figure 11: Definition of chromatographic purification method
Doing these experiments at small scale and defining fixed pooling criteria based on the UV signal prevents extensive in-process analytics in the GMP process, which is often not a viable option due to restricted process hold times and cost considerations.
Lonza’s Conjugation Capabilities
To help continue your bioconjugation journey, Lonza offers a wide range of conjugation scales. This includes only a few milliliter reactions to run early development experiments, up to 10 liters non-GMP pilot batches. Pilot runs play an important role in de-risking scale-up, as it mimics the manufacturing process as closely as possible. Further, it addresses significant material needs for activities prior clinical studies, such as formulation development, toxicology studies, and analytical method validation. For manufacturing, hundreds of liters of bioconjugate reaction with kilograms of dry weight conjugate outputs to supply for commercial demand can be offered (Figure 12).

Figure 12: Lonza’s conjugation capabilities can accommodate your journey throughout clinical- and commercial-phase GMP manufacturing.
Our goal at Lonza is to provide the capabilities necessary to overcome the challenges drug developers typically encounter when moving a bioconjugate to the clinic. The toolbox offering helps with selecting the conjugation technology most suitable for your product and each stage of development it will pass in its path to market. Well-designed process development experiments generate the process knowledge needed to control critical quality attributes and de-risk scale-up in the GMP facility. From the earliest development phase, manufacturability and facility fit are considered in order to avoid re-optimization and repetition. Lonza’s integrated offering from early development to commercial manufacturing covers all the necessary building blocks for a bioconjugate while also reducing the number of interfaces and technology transfers, ultimately de-risking and streamlining your bioconjugate journey.
References
- Citeline: Global Clinical Trial Intelligence Solutions. (2021) https://pharmaintelligence.informa.com/resources/product-content/citeline-global-clinical-trial-solutions
- Yamaguchi, S., Kaneko, M., & Narukawa, M. (2021). Approval success rates of drug candidates based on target, action, modality, application, and their combinations. Clinical and Translational Science, 14(3), 1113–1122. https://doi.org/10.1111/cts.12980
- Jackson, D., & Stover, D. (2015). Using the Lessons Learned from the Clinic to Improve the Preclinical Development of Antibody Drug Conjugates. Pharmaceutical Research, 32(11), 3458–3469. https://doi.org/10.1007/s11095-014-1536-7
- Beck, A., Goetsch, L., Dumontet, C., & Corvaïa, N. (2017). Strategies and challenges for the next generation of antibody-drug conjugates. Nature Reviews Drug Discovery, 16(5), 315–337. https://doi.org/10.1038/nrd.2016.26
- Walsh, S. J., Bargh, J. D., Dannheim, et al. (2021). Site-selective modification strategies in antibody-drug conjugates. Chemical Society Reviews, 50(2), 1305–1353. https://doi.org/10.1039/d0cs00310g
- Junutula, J., Raab, H., Clark, S. et al. (2008). Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 26, 925–932. https://doi.org/10.1038/nbt.1480
- Beacon Targeted Therapies. (2021) www.beacon-intelligence.com