
Navigating The FDA's Emergency Use Authorization Process
By Bikash Chatterjee, president and CSO of Pharmatech Associates, a USP Company and USP Microbiology
On Jan. 31, 2020, the Secretary of Health and Human Services (HHS) declared a national emergency to combat the COVID-19 pandemic caused by the novel coronavirus SARS-CoV-2. As a result, the FDA is authorized to grant Emergency Use Authorizations (EUAs) based upon the criteria defined in its 2017 guidance1 for any medical countermeasure (MCM) that facilitates the diagnosis, prevention, or treatment of SARS-CoV-2. This will impact both internal manufacturing and the CDMOs that drug sponsors employ. Here’s a detailed look at the impacts and details.
COVID-19 pandemic caused by the novel coronavirus SARS-CoV-2. As a result, the FDA is authorized to grant Emergency Use Authorizations (EUAs) based upon the criteria defined in its 2017 guidance1 for any medical countermeasure (MCM) that facilitates the diagnosis, prevention, or treatment of SARS-CoV-2. This will impact both internal manufacturing and the CDMOs that drug sponsors employ. Here’s a detailed look at the impacts and details.
What Is EUA?
The FDA defines EUA as “an expedited authorization and use of an unapproved product or the off-label use of an already approved product in a declared emergency involving a chemical, biological, radiological, or nuclear (CBRN) agent.”2 These MCMs can include drugs, biological products, and devices that have the potential “to diagnose, treat, or prevent serious or life-threatening diseases or conditions caused by a CBRN agent when there are no adequate, approved, and available alternatives.”
The FDA guidance defines six elements of the EUA process as shown in Figure 1 below:
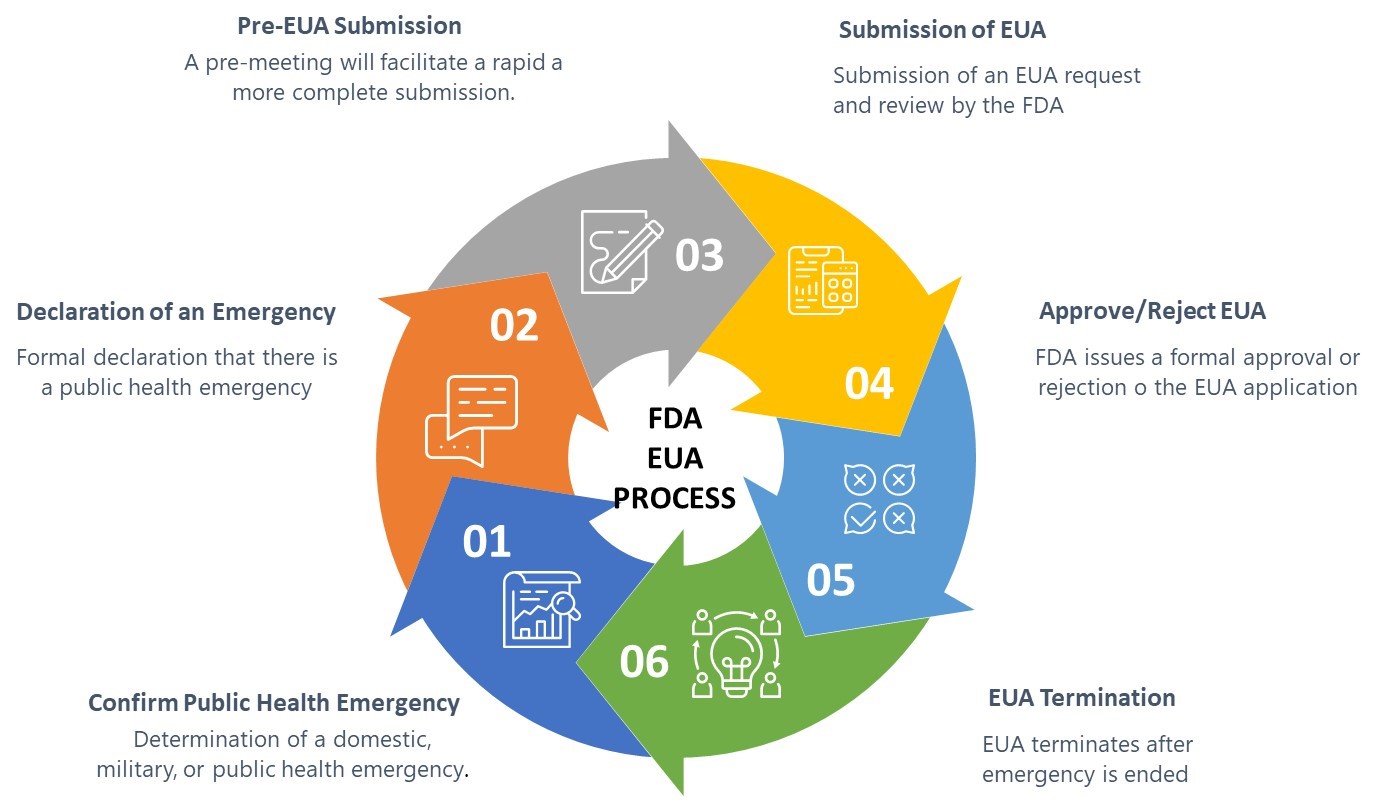
Figure 1: FDA Emergency Use Authorization Process
Satisfying The EUA Criteria
The FDA has established four broad criteria that must be met in order to participate in the EUA program and be granted an EUA:
- Presence of a serious or life-threatening condition
- Evidence of effectiveness
- Risk-benefit analysis (safety)
- No other alternatives to address the life-threatening condition
Presence of Life-Threatening Condition
Specifically, for the FDA to issue an EUA, the CBRN agents referred to in the HHS secretary’s EUA declaration must be capable of causing a serious or life-threatening disease or condition.
Evidence of Effectiveness
For an EUA, the requirement is a lower standard than would be typically applied in a formal regulatory submission, specifically only requiring that the proposed MCM “may be effective” to prevent, diagnose, or treat serious or life-threatening diseases or conditions that can be caused by a CBRN agent identified in the HHS secretary’s declaration of emergency or threat of emergency under section 564(b) of the Federal Food, Drug and Cosmetic (FD&C) Act. The FDA intends to assess the potential effectiveness on a case-by-case basis using a risk-benefit analysis. If, based on the totality of the scientific evidence available, it is reasonable to believe that the product may be effective for the specified use, the FDA may authorize its emergency use, provided that other statutory criteria for issuing an EUA also are met.
Risk-Benefit Analysis
In determining whether the known and potential benefits of the product outweigh the risks, the FDA will look at the totality of the scientific evidence available to make an overall risk-benefit determination. Evidence could arise from a variety of sources available for FDA consideration, such as results of domestic and foreign clinical trials, in vivo efficacy data from animal models, and in vitro data. The FDA recommends that a request for an EUA include a discussion of the candidate product's known and potential risks and benefits, which includes a synthesis of the data and information requested above, including:
- Measures taken to mitigate risk or optimize benefit
- Limitations, uncertainty, and data gaps
- A description of circumstances, if any, under which the product should not be used (e.g., contraindications)
- To the extent known, information concerning the threats posed by the CBRN agents (actually or potentially) involved, and any anticipated response and operational considerations that may be relevant to an assessment of risks and benefits.
While the FDA is intentionally proposing to relax the typical quality framework for product development and manufacturing under cGMPs, it is still requiring verification of effectiveness and safety. For example, ventilator manufacturers are still required to demonstrate their compliance with applicable consensus standards. However, the level of compliance demonstration may be open to discussion and is one example of where a pre-EUA submission meeting with the FDA can be very helpful. In fact, the FDA strongly recommends early engagement with the agency about any potential EUA products to facilitate more complete EUA requests and enhance the FDA’s ability to review and ultimately grant the EUA.
Although time is of the essence in a national emergency, rushing ahead and putting patients at increased risk is a paramount concern for the FDA. Generally, the FDA recommends that data submissions for pre-EUA activities follow recommendations for submitting pre-IND, IND, and device presubmissions. In our experience, this has been invaluable in ensuring the FDA is comfortable with the risk mitigation data, especially when submitting supportive data for MCMs that have been developed via nontraditional pathways, such as academia or manufacturers outside the life sciences industry. Specifically, traditional controls such as requiring a prescription waiver for large-scale distribution of a treatment or requiring a formal risk evaluation and mitigation strategy (REMS) program are often discussed and initially justified in these early meetings.
No Other Alternatives
For the FDA to issue an EUA, there must be no adequate, approved, and available alternative to the candidate product for diagnosing, preventing, or treating the disease or condition. A potential alternative product may be considered “unavailable” if there are insufficient supplies to fully meet the emergency need. A potential alternative product may be considered "inadequate" if, for example, there are contraindicating data for special circumstances or populations (e.g., children, immuno-compromised individuals, or individuals with a drug allergy), if a dosage form of an approved product is inappropriate for use in a special population (e.g., a tablet for individuals who cannot swallow pills), or if the agent is or may be resistant to approved and available alternative products.
EUA Submission
The EUA template is available for download from the FDA website. The FDA recommends that a EUA request include a well-organized summary of the available scientific evidence regarding the product's safety and effectiveness, risks (including an adverse event profile) and benefits, and any available, approved alternatives to the product. The exact type and amount of data needed to support the EUA may vary depending on the nature of the declared emergency or threat of emergency and the nature of the candidate product. The FDA may seek additional data and information on a case-by-case basis to ensure that the statutory criteria for issuance of an EUA are met. Specifically, the EUA submission will require:
- A description of the product and intended use. This should include identification of the serious or life-threatening disease or condition for which the product may be effective; where, when, and how the product is anticipated to be used; and/or the population(s) for which the product may be used.
- A description of the product’s FDA approval status, which should include whether the product or intended use is under an investigational application.
- A summary of the unmet need, including identification of any approved alternative products and their availability and adequacy for the proposed use, and the unmet needs the EUA would address.
- Efficacy and safety information, including data from any clinical studies and nonclinical in vivo and in vitro data on any adverse event monitoring requirements.
- An analysis of the risks and benefits, including any measures taken to mitigate risk or optimize benefit, any contraindications, and any gaps in data. (This analysis should be derived via a structured risk analysis process or framework designed to have the appropriate resolution for the intended use of the MCM.)
- Information on the chemistry, manufacturing, and controls of your product, and on any available and approved alternatives to your product.
- A fact sheet providing information on dosing, contraindications, warnings, and adverse events for distribution to healthcare workers and authorized dispensers of your product.
The timeline for review and approval will vary depending upon the urgency of the need addressed by the submission to a great extent and on the completeness of the EUA submission to mitigate potential risks. The FDA has also established priorities for its review of EUA requests, as described in the guidance.
EUAs Expire With The Emergency
It is important to realize that an MCM approved under a EUA does not signify FDA approval, licensure, or clearance. Upon termination of an EUA as a result of the termination of the HHS EUA declaration supporting it, an unapproved product or its labeling, and product information for an unapproved use of an approved product, must be disposed of pursuant to section 564(b)(2)(B) and (b)(3)59 of the FDA&C Act. The FDA encourages sponsors to continue to develop their products, working toward FDA approval after the termination of the emergency.
Conclusion
The rules for navigating the EUA process are not that different than the principles for a successful regulatory submission. Establishing a strong and clear dialog with the FDA early on will ensure applicants are able to address the primary risks for the intended use of the MCM. Recognizing that the EUA is not a free pass is important, especially for MCMs developed via nontraditional methods. Understanding the FDA’s expectations will allow sponsors to make the most of available data derived during the EUA and potentially position them for a smoother regulatory process once the EUA is revoked. For MCM developers looking to leverage a contract service provider, it is extremely important that both the quality agreement and the supply agreement are clear on critical issues such as intellectual property. This point is especially critical for developers that are new to the marketplace and the liability associated with products not manufactured under a classical cGMP framework.
References:
- Emergency Use Authorization of Medical Products and Related Authorities: Guidance for Industry and Other Stakeholder (Office of Counterterrorism and Emerging Threats) https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization
- https://www.fda.gov/medical-devices/emergency-situations-medical-devices/emergency-use-authorizations
About The Author:
 Bikash Chatterjee is chief operating and science officer for Pharmatech Associates. He has over 30 years’ experience in the design and development of pharmaceutical, biotech, medical device, and IVD products. His work has guided the successful approval and commercialization of over a dozen new products in the U.S. and Europe. Chatterjee is a member of the USP National Advisory Board and is the past chairman of the Golden Gate Chapter of the American Society of Quality. He is the author of Applying Lean Six Sigma in the Pharmaceutical Industry and is a keynote speaker at international conferences. Chatterjee holds a B.A. in biochemistry and a B.S. in chemical engineering from the University of California at San Diego.
Bikash Chatterjee is chief operating and science officer for Pharmatech Associates. He has over 30 years’ experience in the design and development of pharmaceutical, biotech, medical device, and IVD products. His work has guided the successful approval and commercialization of over a dozen new products in the U.S. and Europe. Chatterjee is a member of the USP National Advisory Board and is the past chairman of the Golden Gate Chapter of the American Society of Quality. He is the author of Applying Lean Six Sigma in the Pharmaceutical Industry and is a keynote speaker at international conferences. Chatterjee holds a B.A. in biochemistry and a B.S. in chemical engineering from the University of California at San Diego.