
Essentially Free: How A Definition Tamed The Visible Particulate Matter Debate

By Louis Garguilo, Chief Editor, Outsourced Pharma

In 2005, Stephen Langille became the FDA liaison to the USP Parenteral Products - Industrial Expert Committee. Around that time he and others throughout the drug development and manufacturing industry were becoming concerned with the number of injectable drug product batches rejected or recalled because of issues with “visible particulate matter.”
Part of the matter, if you will, lie within a definition.
“It seemed to stem from the fact USP Chapter <1> – the title at the time was actually “injections” – stipulated each batch of drug product had to be ‘essentially free’ of visible particulates. No one knew exactly what that meant,” recalls Langille. “There wasn’t a good definition for ‘essentially free,” or guidelines as to what was an ‘acceptable level’ of visible particulate matter in injectable drug products.”

But help was on the way.
The USP had published Chapter <790>, which according to Langille, “proved a positive development for both drug manufacturers and the FDA.”
Langille has since left the FDA, but remains an important voice in this segment of our industry. After a 19-year career at the agency, last year he became the Senior Microbiology Consultant at ValSource, LLC.
He’s now released a paper titled, “A Holistic Approach To Visible Particulate Matter Control,” worth reading for any drug developer – perhaps particularly to any employing CDMOs, where many of the visible particulate debates occur.
He offers some advice broadly centered on these questions:
Overall: When to recall and when not to recall?
Process related: What is an acceptable level of contamination within a specific drug product?
Patient focused: How harmful – if at all – are the physical particulate contaminants to the patient?
“That's really what USP <790>, and subsequently USP <1790>, have tried to do,” he explains. “Give the industry, the contract manufacturers, and the regulators a decent amount of background on what the risks are, what the inspection methods should be, and what are acceptable levels of visible particulate contamination for a given lot of drug products.”
And for reader’s visual inspection … here’s a chart from Langille’s newest paper.
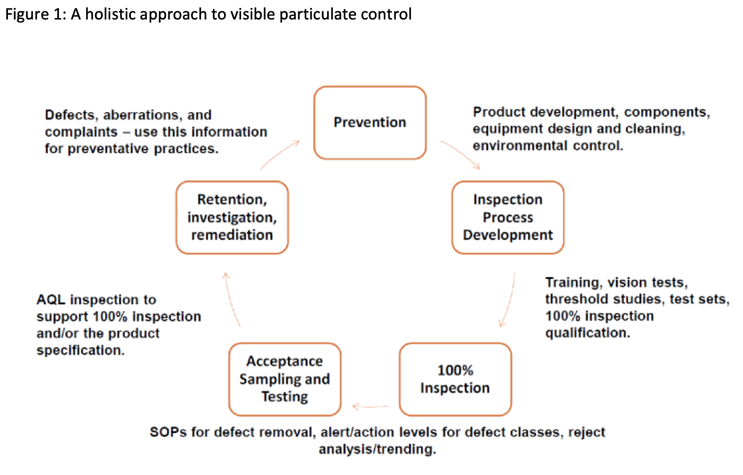
The Definitions
“Regarding the definition of “essentially free” as it pertains to visible particulates in injectable products, my recommendation for your CDMOs is to have them follow the USP recommendations when it comes to establishing a test method and acceptance criteria for visible particulate testing,” Langille advises, and then briefly summarizes those recommendations for us:/p>
USP Chapter <1> says, “The inspection process should be designed and qualified to ensure every lot of all parenteral preparations is essentially free from visible particulates, as defined in Visible Particulates in Injections <790>.”
USP <790> establishes a test method and minimum acceptance criteria in the form of an acceptable quality limit (AQL) for a batch of drug product that has already been subjected to a 100% inspection for visible particles and other defects. The visible particle test method and acceptance criteria may be included in the regulatory submission in order to gain regulatory approval for the visible particle control strategy. The acceptance criteria listed in USP <790> is consistent with that of a major defect, one that would affect the usability of the product but not a threat to patient safety. A critical defect, on the other hand, would be one that would result in hazardous or unsafe conditions for those using it.
Got all that?
Here’s Langille’s elemental checklist/questions for what drug product owners should ensure is established or should be asked of a CDMO’s visual inspection program.
- Threshold studies should be conducted by either the product owner or the CDMO to establish the size range of different types of particles that are visible in the product.
- Are operators qualified with adequate blinded test sets and do they undergo regular vision exams?
- Is the inspection station free of distraction and properly maintained (e.g., black and white background free of scratches and defects).
- Is the 100% inspection process qualified to detect visual defects and visible particles under the conditions of use? This is especially important for semi-automated visual inspection processes which often have throughputs of >1 unit per second.
- Is the manufacturing environment, components, container system, equipment, and facility adequate to prevent the introduction of visible particles into the finished injectable product? Prevention of contamination is the most important part of visible particulate control.
I asked Langille if there’s in fact a way to discern a priori competency at CDMOs in these areas?
“I would make sure the entire visual inspection processes has been validated, and the operators trained and qualified,” he replies. “Biopharma companies should ask what specific inspection process is being used – for example, European Pharmacopoeia chapter 2.9.20 or USP chapter <790> -- and what their acceptance criteria are for the 100% inspection and quality test.
“Ask how the operators have been qualified. Are regular vision tests being conducted? Are there blinded-qualification studies where operators are asked to identify visible particulates in a test set? Inspections can be checked for the ability to identify physical defects.
“All this should be investigated closely by any drug owner of their CDMO.”
Speaking As Important As Seeing
Avoiding problems often calls into play yet another factor.
“Over my career, I have found that usually problems stem from a lack of communication between the customer and the CDMO,” says Langille.
“For instance, if a CDMO is making a large number of products – with varying container sizes, shapes, orientations from ampules to vials, prefilled syringe or even blow-fill-seal containers – some of those products might be familiar to the CDMO, but the visual inspection processes and nuances of those specific products and enclosure systems should be discussed in detail. There can be differences in product colors, opacity, and even the potential for inherent particular matter that could affect the visual inspection process.
“And this is especially true with traditional biologics and advanced therapy medicinal products,” cautions Langille. “CDMOs should be made aware of that.”
“I've seen cases where biological products had inherent particulate matter – harmless byproducts as part of the manufacturing process – that looked like fibers. With one product, these inherit particulates were misidentified as extrinsic fiber contaminants during a CDMO’s inspection. The CDMO rejected about half of the batch. It just hadn’t been clearly communicated to the CDMO by the biopharma customer. Nuances of the product affecting inspection must be straightened out ahead of time between the CDMO and the license holder.”
The Scrutinized CDMO
Langille offers a final thought.
“There will always be scrutiny on the CDMOs, simply for the fact they are not the drug owners. They are subjected to a lot of attention because they're making different products at their facilities – they have to prove expertise in every single one.
“That’s difficult for anyone, CDMO or otherwise. And CDMOs at times are ‘put through the ringer’ inspection-wise by some of their customers, and the various regulatory agencies around the world.
“And nowadays CDMO are asked to manufacture an even wider variety of products, with more complex container-closure systems … I think that's a real challenge.
“I feel for them,” concludes Langille. “It’s not an easy gig.”