
What 2025 FDA Warning Letters Tell Us About GMP Compliance
By Ajay Babu Pazhayattil, Marzena Ingram, and Koti Reddy Bhimavarapu

This analysis examines the 2025 U.S. FDA warning letters issued to drug product manufacturers and posted between January 1 and December 9, 2025, identifying key compliance trends and regional disparities. The review of 85 letters reveals not only the most frequently cited regulatory deficiencies but also significant geographical patterns in the nature of violations, particularly concerning data integrity. By mapping the most frequently cited sections of 21 CFR Part 211, the analysis highlights where quality systems are breaking down and where proactive remediation efforts should be focused. Fifty-nine percent of the warning letters were issued to U.S. facilities, followed by sites in China, India, Canada, and Turkey.

Figure 1: 2025 Drug Product Site Warning Letters
A particularly revealing insight emerges from the data integrity (DI) findings. While 15% of all warning letters reviewed cited DI concerns, they are not evenly distributed. Indian sites received warning letters at a 60% rate, with associated DI issues, far surpassing that of sites in the U.S. (10%) and China (21%). This significant disparity suggests that data integrity may represent a systemic challenge within certain regional manufacturing landscapes, potentially reflecting differences in quality culture. Furthermore, the FDA's recommendation for a GMP consultant in 87% of the letters is a striking commonality. This nearly universal point emphasizes that the observed failures are often not isolated incidents but indicative of broader embedded weaknesses in quality management systems that require external consultant expertise to adequately remediate non-compliance scenarios. Notably, over-the-counter (OTC) manufacturers, as identified by the FDA, received 11 % of all warning letters.

Created with Datawrapper
Figure 2: Percentage of Warning Letters Citing Data Integrity Issues
The top 21 CFR Part 211 GMP sections discussed in the warning letters are 84(d)1, 2; 22(a), (d); 100(a); 165(a); and 192.
- Sections 211.84 (d)1 and (d)2 pertain to testing and approval or rejection of components, drug product containers, and closures. Subsection (d)1 requires that at least one test be conducted to verify the identity of each component of a drug product, and subsection (d)2 requires that each component be tested for conformity with all appropriate written specifications for purity, strength, and quality.
- Section 211.22 is on the responsibilities of the quality control unit. Notably, 22(a) requires that a quality control unit has the responsibility and authority to approve or reject all components, drug product containers, closures, in-process materials, packaging material, labeling, and drug products and the authority to review production records to assure that no errors have occurred or, if errors have occurred, that they have been fully investigated. The quality control unit shall be responsible for approving or rejecting drug products manufactured, processed, packed, or held under contract by another company. Section 22(d), on the other hand, clarifies that the responsibilities and procedures applicable to the quality control unit are written and followed.
- Section 100 (a) discusses the need for written procedures for production and process control designed to assure that the drug products have the identity, strength, quality, and purity they purport or are represented to possess. Note that this section pertains to GMP regulations regarding lifecycle-based process validation (PV Stages 1, 2 and 3).
- Section 211.165 on testing and release for distribution states that, for each batch of drug product, there are appropriate laboratory determinations of satisfactory conformance to final specifications for the drug product, including the identity and strength of each active ingredient, prior to release.
- Section 211.192 on production record review requires that all drug product production and control records, including those for packaging and labeling, be reviewed and approved by the quality control unit to determine compliance with all established, approved written procedures before a batch is released or distributed. Any unexplained discrepancy (including a percentage of theoretical yield exceeding the maximum or minimum percentages established in master production and control records) or the failure of a batch or any of its components to meet any of its specifications is thoroughly investigated, whether or not the batch has already been distributed. The investigation is to be extended to other batches of the same drug product and to other drug products that may have been associated with the specific failure or discrepancy. Therefore, conducting a scientifically sound and comprehensive investigation, supported by a robust root-cause analysis that drives effective corrective and preventive actions, is critical.
The most frequently cited violation was 21 CFR 211.84(d)(1), pertaining to identity testing of components, cited in 49 warning letters. This was closely followed by 211.84(d)(2) on testing for purity, strength, and quality, cited in 41 warning letters. Together, the significant takeaway is that companies should prioritize a global audit of their supplier qualification and incoming material control systems. Other significant deficiencies include failures to establish and follow validated procedures (211.100(a), 33 citations), inadequate laboratory testing before batch release (211.165(a), 26 citations), and insufficient production record review and investigation (211.192, 22 citations). Underlying these specific lapses are broader quality system weaknesses, including inadequate authority and defined responsibilities for the quality unit (211.22, 211.22(a), and 211.22(b), with a total of 54 citations).
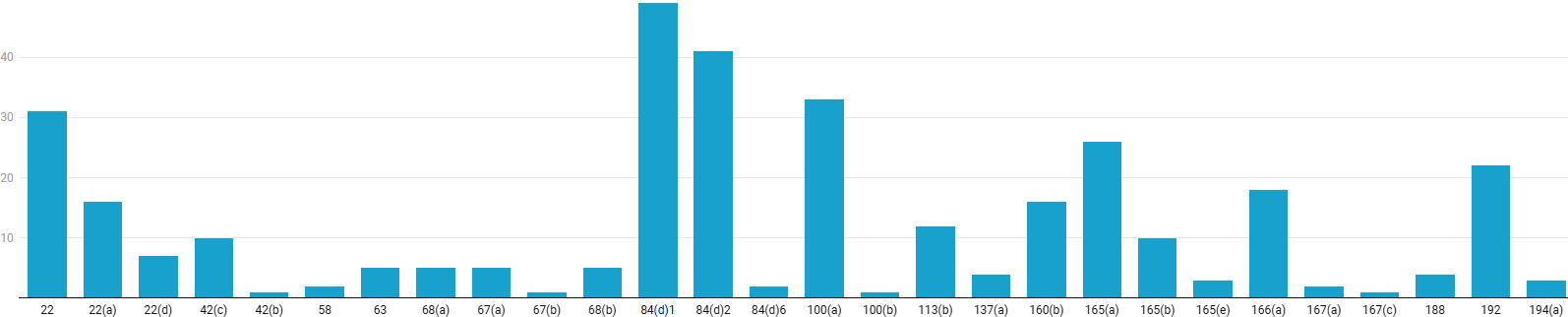
Created with Datawrapper
Figure 3: GMP Sections Cited in Warning Letters. Click on the image to enlarge.
The 2025 FDA warning letter data reveal several insights. The known vulnerabilities in quality unit performance, testing, process validation, and investigations continue to be a hurdle for manufacturers. Profound regional disparities exist, most notably in data integrity compliance, with Indian facilities facing disproportionately high rates, indicating challenges in data governance and oversight culture. The overwhelming frequency with which the FDA recommends third-party GMP consultation indicates that many companies lack the internal capability to self-correct these systemic issues. The analysis shows that systemic weaknesses in supplier qualification and the incoming testing program can result in warning letters. Targeted plans to address regulatory vulnerabilities are essential to mitigate risks, given the recurring compliance failures that have led to warning letters.
About The Authors:
 Ajay Babu Pazhayattil, Ph.D., is an accomplished management consultant and industrial pharmacist with extensive experience spanning solid oral, sterile, and API sectors. He is the founder of cGMP World. Pazhayattil has held key leadership positions with prominent North American brands, generic manufacturers, and CDMOs. His roles include vice president of scientific and regulatory affairs at Capcium, quality director at Eurofins, and associate director at Apotex. He plays a pivotal role in guiding organizations through remediation efforts related to U.S. FDA 483 observations and warning letters. He has been a lead author and contributor to guidance documents published by industry organizations such as AAPS, PDA, ISPE, and RAPS.
Ajay Babu Pazhayattil, Ph.D., is an accomplished management consultant and industrial pharmacist with extensive experience spanning solid oral, sterile, and API sectors. He is the founder of cGMP World. Pazhayattil has held key leadership positions with prominent North American brands, generic manufacturers, and CDMOs. His roles include vice president of scientific and regulatory affairs at Capcium, quality director at Eurofins, and associate director at Apotex. He plays a pivotal role in guiding organizations through remediation efforts related to U.S. FDA 483 observations and warning letters. He has been a lead author and contributor to guidance documents published by industry organizations such as AAPS, PDA, ISPE, and RAPS.
 Marzena Ingram is an independent senior pharmaceutical consultant with extensive QA, technical operations, and process validation expertise. Her specialty is in navigating FDA warning letter scenarios and providing clients with regulatory-compliant solutions. She is known for strategic team leadership. She has driven compliance initiatives that have met global regulatory standards and has been involved in establishing industry benchmarks. Additionally, Ingram has built and led specialized teams in high-volume manufacturing settings, ensuring process validation excellence and performance. Ingram has served as VP of ISPE Canada.
Marzena Ingram is an independent senior pharmaceutical consultant with extensive QA, technical operations, and process validation expertise. Her specialty is in navigating FDA warning letter scenarios and providing clients with regulatory-compliant solutions. She is known for strategic team leadership. She has driven compliance initiatives that have met global regulatory standards and has been involved in establishing industry benchmarks. Additionally, Ingram has built and led specialized teams in high-volume manufacturing settings, ensuring process validation excellence and performance. Ingram has served as VP of ISPE Canada.
 Koti Reddy Bhimavarapu is a pharmaceutical consultant and data integrity subject matter expert with extensive experience supporting compliance-driven organizations. He brings strong cross-functional expertise across quality control and quality assurance operations, cGMP and cGLP management, analytical method validation, stability studies, and overall regulatory compliance. As a GMP consultant, he advises clients in both API manufacturing and finished dosage formulation environments. Koti has previously served in senior quality leadership roles, including head of quality positions at Cipla, Wockhardt, Microlabs, and Dr. Reddy's.
Koti Reddy Bhimavarapu is a pharmaceutical consultant and data integrity subject matter expert with extensive experience supporting compliance-driven organizations. He brings strong cross-functional expertise across quality control and quality assurance operations, cGMP and cGLP management, analytical method validation, stability studies, and overall regulatory compliance. As a GMP consultant, he advises clients in both API manufacturing and finished dosage formulation environments. Koti has previously served in senior quality leadership roles, including head of quality positions at Cipla, Wockhardt, Microlabs, and Dr. Reddy's.