
Reliably Predicting Biologics Hotspots From Prior Knowledge
By Ramakrishnan Natesan and Neeraj J. Agrawal, Amgen

In 2024, the 900-plus FDA-approved protein-based therapeutics (PBTs) constituted a market share of $385 billion and is one of the fastest growing segments in the drug industry, expected to grow at 6.3% annually to $550 billion in 2031.1,2 PBTs, also known as “biologics,” commonly refer to therapeutic molecules such as monoclonal antibodies (mAbs), recombinant proteins, antibody-drug conjugates, fusion proteins, peptides, and other modalities containing naturally occurring amino acids.
This approach, wherein we engineer therapeutic function(s) into native amino acid sequences, offers multiple benefits over other modalities. PBTs are known to bind to their targets with high specificity, high affinity, and display desirable pharmacokinetics with longer circulation times. Furthermore, the immense growth in biotechnology to engineer protein-based therapeutics with a wide spectrum of target-engagement strategies and mechanisms of action has enabled therapeutic development against targets that were once deemed "undruggable."
The quality of a biologic molecule is strongly dependent on the molecule's critical quality attributes (CQAs). As defined in ICH Q8 (R2)3, a CQA is “a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality.”
Site-specific post translational modifications (PTMs) in a biologic molecule are CQAs known to significantly impact quality due to their ability to influence the molecule’s structure and function. These site-specific modifications are commonly referred to as chemical hotspots/liabilities. Well known examples of chemical hotspots include asparagine/glutamine deamidation, aspartic acid isomerization, tryptophan/methionine oxidation, glycation of lysine, loss of C-terminal lysine, and pyroglutamate formation of N-terminal Glu/Gln. 4
The presence of such undesired modifications could impact the safety and/or efficacy of the molecule. Hence, an early identification or prediction of these chemical hotspots is desirable to either design strategies to optimize the protein sequence for hotspot remediation or to develop CMC processes for robust control of these hotspots.
Identification Of Chemical Hotspots From Prior Knowledge
Conventionally, a tour de force approach is applied to de-risk hotspots in a molecule. Samples are subjected to a specific external stress, and the abundances of all eluted species are quantified using LC-MS/MS peptide mapping. Generating a complete panel of molecular hotspots requires a large set of such experiments covering a wide range of controls and stress conditions, which in turn requires substantial resources and time. Hence, such assays are generally performed during later phases of the CMC development process.
Alternatively, presence of potential chemical hotspots can be predicted using computational approaches (e.g., motif detection, local flexibility, solvent accessibility, and hydrogen bonding) or through a similarity approach using prior knowledge from historical peptide mapping data generated for previous programs of similar modality.
In this article, we describe a data-driven workflow we have developed at Amgen for rapidly de-risking chemical hotspots. This approach allows us to quickly filter out low propensity/abundance hotspots and thus enable prioritization of high abundance chemical hotspots for further experimental assessment. This workflow utilizes the vast trove of peptide mapping data generated as part of all our programs to date.
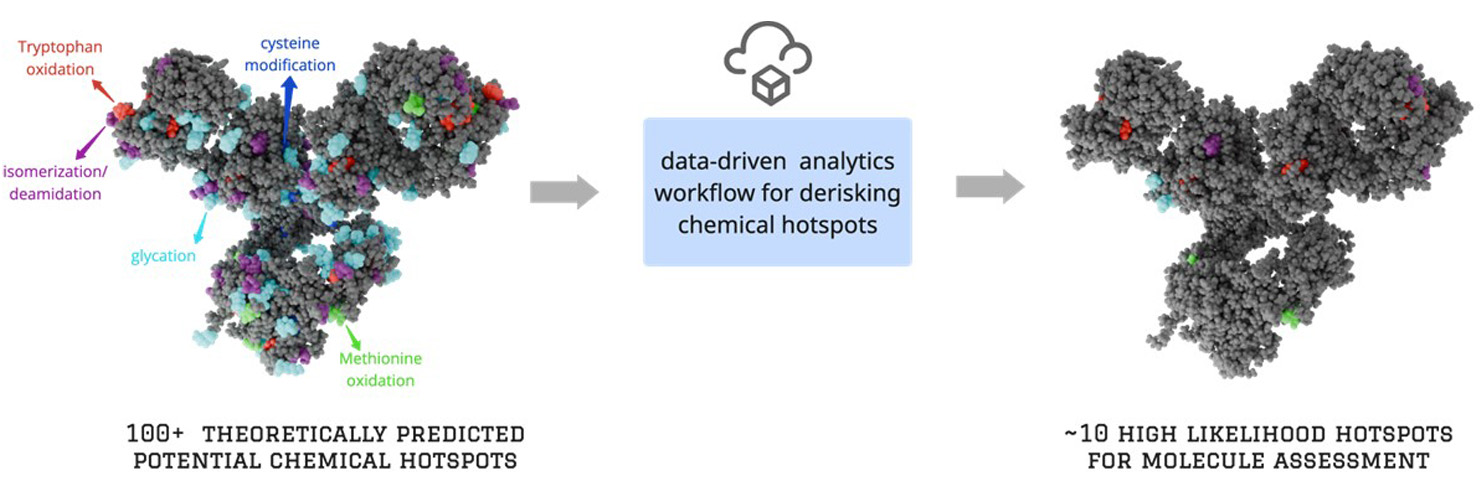
(Click image to view full size)
Figure 1: Illustration of our automated workflow for de-risking hotspots. The left panel shows the full list of predicted hotspots for an IgG1 mAb, while the right panel displays only those predicted to have a high propensity for PTM.
We first digitized internal peptide mapping data sourced from our electronic notebooks and various legacy systems and built a standardized database (PePMapDB) containing the abundance of site-specific PTM in mAbs and mAb-fragment containing molecules. This data set covers hundreds of internal programs across various stages of drug development (from discovery to commercial) and provides detailed insight into a molecule’s attribute under native and stress conditions. 5,6 The data was standardized by aligning all molecules to an IgG-specific reference sequence and numbered using an internal numbering system that closely follows a published numbering scheme. 7 A technical description of the database building effort may be found in the article by Jacobitz, Rodezno, and Agrawal. 8
We also collected sequence information for all commercial mAbs and generated an additional database (CommercialmAbDB) with identical alignment and numbering to construct an additional layer of statistics for de-risking. The underlying hypothesis for this additional step is: A hotspot that is part of multiple commercial molecules tends to pose a lower risk since these hotspots have been thoroughly evaluated for developability and risk and found to be either benign or could be mitigated with available manufacturing and/or storage conditions.
These prior knowledge databases can be reliably used to evaluate the risk associated with a site-specific hotspot. Internally, we have implemented this workflow as a web app, a schematic of which is shown in Figure 2. The app accepts a user-uploaded sequence alongside user-defined cutoffs for classification as inputs and provides a detailed list of risky and non-risky hotspots, with the bulk of the potential hotspots falling in the latter category, as illustrated in Fig. 1. All hotspots marked risky are automatically flagged for subsequent triage and experimental verification.
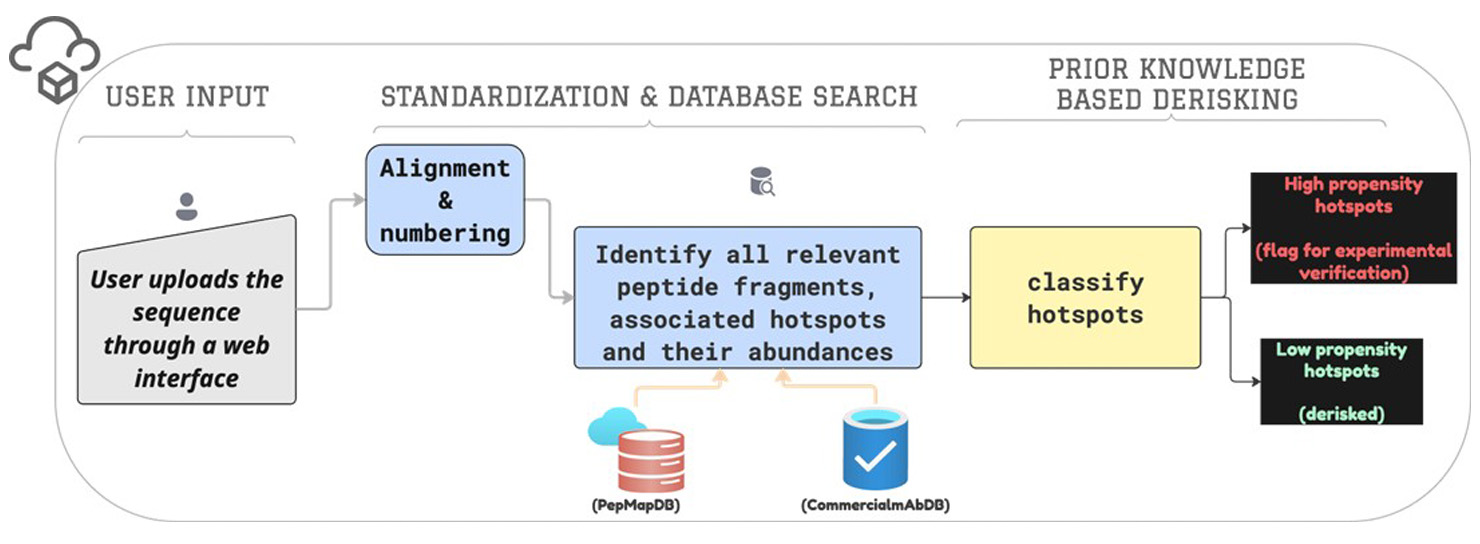
(Click image to view full size)
Figure 2: An overview of our automated workflow for hotspot de-risking.
Case Study: De-risking Hotspots For The NISTmAb
We will next demonstrate the utility of our approach by de-risking hotspots associated with NISTmAb RM 8671, an IgG1![]() type research monoclonal antibody. In the initial prediction step (referred to as Stage 1 in Fig. 3), our workflow predicts 118 potential chemical hotspots distributed across 12 different PTMs. In Stage 2, we identified all sites not found in at least 50% of commercial mAbs and late-stage assets (i.e., occurrence frequency (OF) < 50%), and this filtering step eliminated nearly three-fourths of the predicted hotspots, bringing the number of potential chemical hotspot sites to 34. In the center panel in Fig. 3, the vertical dashed line corresponds to OF=50%, points colored green correspond to de-risked hotspots, and points colored red correspond to risky hotspots that progress to the next step of evaluation.
type research monoclonal antibody. In the initial prediction step (referred to as Stage 1 in Fig. 3), our workflow predicts 118 potential chemical hotspots distributed across 12 different PTMs. In Stage 2, we identified all sites not found in at least 50% of commercial mAbs and late-stage assets (i.e., occurrence frequency (OF) < 50%), and this filtering step eliminated nearly three-fourths of the predicted hotspots, bringing the number of potential chemical hotspot sites to 34. In the center panel in Fig. 3, the vertical dashed line corresponds to OF=50%, points colored green correspond to de-risked hotspots, and points colored red correspond to risky hotspots that progress to the next step of evaluation.
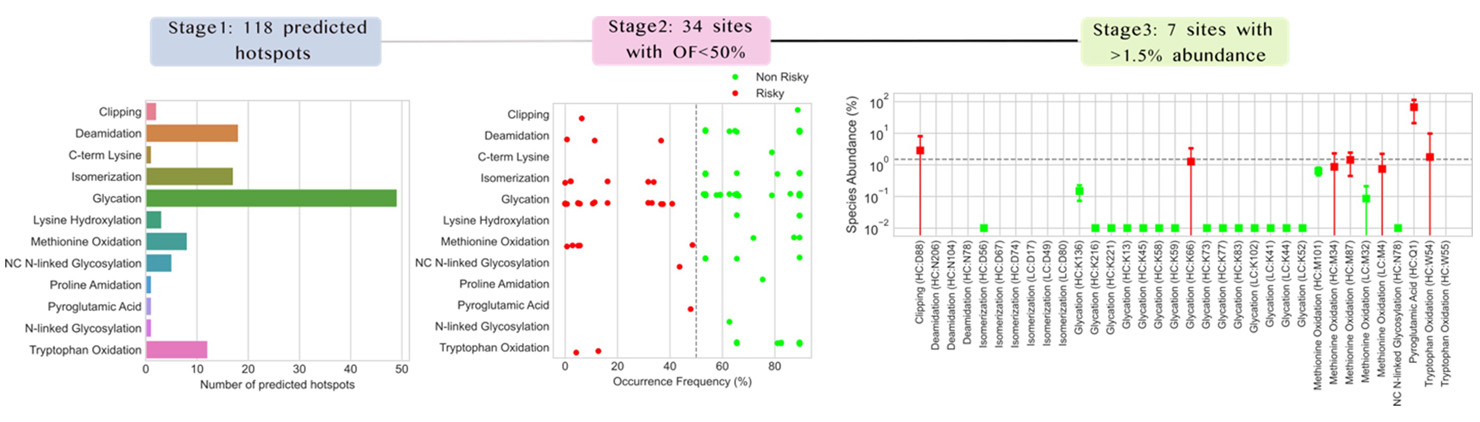
(Click image to view full size)
Figure 3: Results of our workflow applied to the NISTmAb RM 8671 sequence. The left panel displays the PTM specific distribution of the 118 theoretically predicted hotspots. The center panel shows the occurrence frequency (OF) of each chemical hotspot in commercial mAbs and late-stage assets. Hotspots with OF>=50% are colored green while the 34 hotspots with OF<50% are colored red. In the right panel, experimentally measured abundances for each of the 34 hotspots are displayed as a boxplot. Here the abundance score threshold (horizontal dashed line) was set to 1.5% and hotspots exceeding the threshold are colored red while those below the threshold are colored green. Our analysis yields just seven sites for further experimental validation.
In Stage 3 of the de-risking workflow, we evaluate the abundance levels of these 34 hotspots in prior peptide mapping data and retain all sites with species abundance > 1.5%. Much of the peptide fragments not detected in commercial mAbs were also not detected in early molecule assessments, thus bringing the final list of risky hotspots to just seven, which are recommended for assessment with further experimental studies. In this example, we have chosen a stringent abundance cutoff of 1.5%, but this threshold can be adjusted based on internal guidelines and risks associated with a specific PTM.
Conclusions
We have presented an automated data-driven workflow that uses publicly available molecular sequence data and proprietary peptide mapping data to reliably de-risk chemical hotspots in mAbs. This data-driven approach is highly robust, extensible, and the predictions only get better as the underlying data corpus grows. As with any data-driven platform, the validity of the tool entirely depends on the quality of the underlying data and, hence, extra care should be taken in the data curation step.
Our automated workflow tool has enabled our lab scientists to rationally select appropriate molecular attributes for assessment, which has helped us realize substantial savings in resources and time. In our experience, this tool has helped our teams in advancing timelines for multiple programs.
References:
- Jain, S., Gupta, S., Patiyal, S. & Raghava, G. P. S. THPdb2: compilation of FDA approved therapeutic peptides and proteins. Drug Discovery Today 29, 104047 (2024).
- Protein Therapeutics - Global Strategic Business Report. Global Industry Analysts, Inc (2025).
- International Conference On Harmonisation Of Technical Requirements For Registration Of Pharmaceuticals For Human Use. in Handbook of Transnational Economic Governance Regimes 1041–1053 (Brill | Nijhoff, 2010). doi:10.1163/ej.9789004163300.i-1081.897.
- Chiu, M. L., Goulet, D. R., Teplyakov, A. & Gilliland, G. L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies (Basel) 8, (2019).
- ICH HARMONISED TRIPARTITE GUIDELINE. QUALITY OF BIOTECHNOLOGICAL PRODUCTS: STABILITY TESTING OF BIOTECHNOLOGICAL/BIOLOGICAL PRODUCTS Q5C. https://database.ich.org/sites/default/files/Q5C%20Guideline.pdf (1995).
- ICH HARMONISED TRIPARTITE GUIDELINE. STABILITY TESTING OF NEW DRUG SUBSTANCES AND PRODUCTS Q1A. https://database.ich.org/sites/default/files/Q5C%20Guideline.pdf (2003).
- Dondelinger, M. et al. Understanding the Significance and Implications of Antibody Numbering and Antigen-Binding Surface/Residue Definition. Front Immunol vol. 9 2278 (2018).
- Jacobitz, A. W., Rodezno, W. & Agrawal, N. J. Utilizing cross-product prior knowledge to rapidly de-risk chemical liabilities in therapeutic antibody candidates. AAPS Open 8, 10 (2022).
About The Authors:
 Ramakrishnan Natesan, Ph.D., is a principal data scientist at Amgen who works in the areas of computational physics, soft condensed matter, biophysics, bioengineering, and fluid mechanics. His work focuses on applying multiscale modeling and machine learning for accurate characterization of molecular attributes and early-stage optimization of biologics. Prior to joining Amgen, he conducted postdoctoral work at the University of Pennsylvania. He received his Ph.D. in physics from the Indian Institute of Technology Madras.
Ramakrishnan Natesan, Ph.D., is a principal data scientist at Amgen who works in the areas of computational physics, soft condensed matter, biophysics, bioengineering, and fluid mechanics. His work focuses on applying multiscale modeling and machine learning for accurate characterization of molecular attributes and early-stage optimization of biologics. Prior to joining Amgen, he conducted postdoctoral work at the University of Pennsylvania. He received his Ph.D. in physics from the Indian Institute of Technology Madras.
 Neeraj Agrawal, Ph.D., is a senior director in process development at Amgen where he leads the pivotal attribute/analytical sciences team overseeing late-stage product characterization. Previously, he worked as a scientist at Ipsen. He received his Ph.D. in chemical engineering from the University of Pennsylvania and did postdoctoral work at MIT.
Neeraj Agrawal, Ph.D., is a senior director in process development at Amgen where he leads the pivotal attribute/analytical sciences team overseeing late-stage product characterization. Previously, he worked as a scientist at Ipsen. He received his Ph.D. in chemical engineering from the University of Pennsylvania and did postdoctoral work at MIT.