
Integrating Excipients Into QbD For Drug Development: Understanding Drift & Critical Material Attributes
By Brian Carlin (DFE Pharma), Chris Moreton (FinnBrit Consulting), Dave Schoneker (Black Diamond Consulting), Katherine Ulman (KLU Consulting), Joseph Zeleznik (IMCD)

This article is the second in a three-part series based on information from the new guide Incorporation of Pharmaceutical Excipients into Product Development using Quality-by-Design, published by the International Pharmaceutical Excipients Council Federation (IPEC Federation).1 Click here to read Part 1.
Product, Process, And Excipient Drift
Proximity to percolation thresholds, failure points, or balance point between conflicting requirements will predispose a product to being impacted by excipient variability. During development, the variability of a basic2,3 excipient (or other attributes of a performance excipient) may be far enough away from finished product criticalities so as not to elicit a response under the experimental conditions. The question then arises: How can so-called “non-critical” excipients be associated with special cause variation at some stage in the product life cycle? Their impact is indirect, due to interaction with finished product criticalities. If no such interactions were observed during development, then something has changed, resulting in product drift. This could include the cumulative impact of API, equipment, excipient, and/or process changes, as shown below.

Such drift may not be detectable by univariate change control, hence the value of continued multivariate monitoring. Pharmaceutical product change control is nearly always univariate. Product performance is checked before and after the change to confirm that the product remains within specification and that the change has had no effect on CQAs. This may be formalized as a comparability protocol to cover foreseeable events such as switching excipient suppliers. The weakness of this approach is that other product attributes may change, which are not reflected in specification parameters. After several supplier changes and process adjustments, sequentially qualified one step at a time, the system may have drifted, and the next change triggers an unexpected problem. Past performance is not predictive of future performance. Ideally, tracking of excipient attributes and finished product CQAs should be complemented with multivariate monitoring.
Note: The presenting excipient variability may not be causative. Excipients and their variability may not have changed but are now within range of a criticality and starting to correlate with finished product quality excursions.
Impact from drift can be illustrated by simple process capability models as shown in Figure 1.
Process capability is essentially the ratio of specification range relative to range of variability, usually quantified as ±3 standard deviations (±3σ), assuming only common cause variation. It is a measure of the ability of the process to yield product within limits. For the purposes of illustrating the combined impact of drift, variability, and criticality, only a single limit is needed. This corresponds to an unknown limit within the product beyond where excipient variability will impact product CQAs.
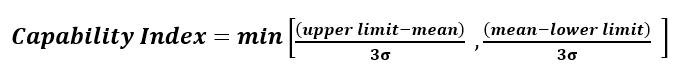
Figure 1: Simple Process Capability Model
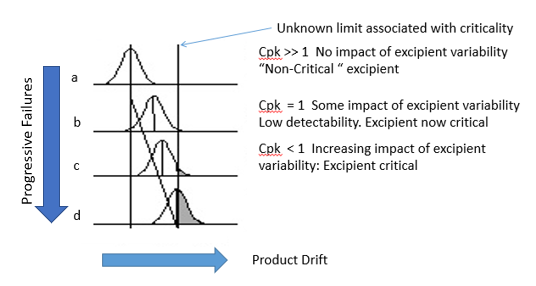
In Figure 1a, the criticality limit is on the right-hand side. The impact of excipient variability is not seen as it is well to the left of the criticality limit. The process capability is >1, and the product can tolerate some drift, with the 3σ limit being well away from the criticality. The excipient would be regarded as non-critical, as there is no discernible impact on product CQAs.
Figure 1b shows the 3σ limit drifting to the criticality limit. The process capability now has a value of one, and the previously non-critical excipient is now critical. Excipient variability is starting to correlate with product quality excursions. Detectability is low, as incidence of excursions will still be parts per million, assuming a normal distribution. Further drift (Figures 1c and 1d) increases the incidence of quality excursion, and the process is no longer capable of producing product within limits.
Note: Excursion severity may be disproportionate if drift straddles a percolation threshold between two different regimes.
The level of effort, formality, and documentation of the quality risk management process should be commensurate with risk level. Binary excipient categorization into critical versus non-critical has been criticized as being overly simplistic and not consistent with science and risk-based thinking. It is better to rank excipient criticality with the relative risk of not achieving desired quality attributes, a “spectrum of importance”4 with respect to material attributes. All excipients are critical, but at a given time, some have more impact in an application than others. A suggested approach is to categorize excipients as either performance or basic, using an adaptation of Kano Analysis.2, 3
- Performance excipients have one or more attributes that have direct, proportional, and immediate impact on finished product quality. These are what would traditionally be regarded as critical excipients and are titrated into a formulation for specific performance.
- Variability in attributes of basic excipients has no direct or immediate impact on finished product quality, above a minimum threshold. Such excipient attributes are generally taken for granted by users and only result in dissatisfaction if not present or insufficient. It is more appropriate to think of these excipients as potentially critical (below threshold) rather than non-critical.
Critical Material Attributes (CMAs)
A CMA is defined as an attribute of the excipient that must be within appropriate limits to ensure that the CQAs for the drug product are maintained throughout the product life cycle. Alternatively, the CMA may be used to control the process or the level of addition of the excipient (or other formulation components).
A CMA may be an attribute that is not part of the pharmacopeial or supplier specification for the excipient. Note that the CMA is specific to the finished product, not to the excipient. The same excipient used for multiple products may be critical in some but not in others. A CMA is not the raw material equivalent of a CQA.
A CQA of a finished product is an attribute that must be within appropriate limits to ensure the quality of the finished product. By analogy, excipient CQAs are those attributes comprising the excipient specification. They guarantee the quality of the excipient in isolation, but not performance or fitness for purpose in the finished product. If tighter excipient limits than specified are required, or additional excipient attributes need to be specified, in order to maintain finished product quality, then such additional controls are CMAs. In this respect, a CMA is closer to a critical process parameter (CPP) than a CQA.
Product-specific CMAs should also be distinguished from application-specific functionality related characteristics (FRCs). FRCs are potential CMAs but are non-mandatory (Ph. Eur.) or listed without acceptance criteria (USP/NF monograph Labeling section). The impact of FRCs on finished product quality should be evaluated during development. If specific limits are required, they become CMAs. If not critical, limits should not be specified by the user, but the FRCs should be monitored as part of the user’s control strategy (CS).
For example, in solid dose forms, particle size distribution (PSD) is an FRC, which only becomes a CMA if additional or tighter limits need to be imposed in addition to supplier-grade specifications to control the finished product quality.
PSD is irrelevant where excipients are in solution but is likely critical in solid dose forms. Table 1 illustrates the spectrum of criticality for lactose PSD across a range of dosage forms.
Table 1: The Criticality of Lactose Particle Size Distribution in Different Dose Forms
|
Dose Form |
CMA |
FRC |
|
|
Dry Powder Inhaler |
Yes |
Yes |
Product specific, particle size nearly always critical |
|
Solid Dose Form |
Maybe |
Yes |
Application specific, particle size may be critical |
|
Solution |
No |
No* |
Not critical |
* The Ph. Eur. emphasizes that the relevance of the FRC in a particular application is determined by the excipient user.
Excipient attributes that must be controlled to ensure compliance with specification are CQAs for that excipient but are not necessarily CMAs. They are minimum standards with limited relevance to fitness for purpose in specific applications. CMAs are complementary to compliance. Reliance on pharmacopeial compliance alone implies no CMAs are required, but this requires justification for products more complex than simple solutions.
CMAs should be discussed early in development with suppliers to ensure that desired limits, ranges, or distributions are within supplier process capabilities. Suppliers also may be able to suggest other potential CMAs and provide appropriate QbD samples for specific applications.
CMAs are complementary to CPPs in controlling finished product quality. A fixed formula with a fixed process will be entirely dependent on CMAs, as there are no compensatory controls. For example, a granulation process with a target performance endpoint (CPP), such as torque, may not need CMAs for the granulation excipients. However, the same process with a fixed endpoint, such as granulation time, may be dependent on CMAs.
CMAs can be used to either exclude excipient batches outside of required limits or control the process via signals from the attribute(s) of interest. The latter affords greater sourcing flexibility in that wider limits can be used where compensatory process controls exist. Formulation control can also be applied by both user and manufacturer:
- Users can blend grades varying in CMAs to achieve a consistent, desired CMA.
- Manufacturers can supply standardized performance using agreed methods and an agreed diluent (batch-to batch compositional variability, but consistent performance in target application).
CMAs should be ranked in terms of severity of impact on patient safety, followed by severity of impact on manufacturability. Detectability may be a secondary consideration but probably is of limited value in ranking CMAs given the potential for special cause variation, which is inherently unpredictable, even probabilistically. Failure to rank means that a reviewer will be unable to determine the relative importance of the CMAs. If there is a large number of unranked CMAs, unnecessary scrutiny and delay may result, but the number of CMAs should not be minimized for fear of regulatory scrutiny.
CMAs are often performance attributes, which directly and proportionally impact finished product CQAs. For example, polymer attributes that control rate of release may require narrower ranges or distributions (to be determined experimentally) than those allowed by pharmacopeial limits to ensure consistent release profiles.
Performance attributes require little experimental confirmation as they tend to be dominant, having already been titrated into prototype formulations. In addition to desired functionalities, side effects may also prompt CMA assignment. For example, magnesium stearate is intended to improve tablet manufacturability by easing ejection and reducing sticking. In practice, the well-known adverse effects on dissolution and compactability may outweigh functional benefits and require control via CMAs.
It is difficult to determine the criticality of basic attributes, as they may exhibit a no response region above a threshold. Absence of impact under experimental conditions does not rule out potential impact if circumstances change, e.g., during scale-up. Good experimental design using matrix statistical treatments is more likely to identify interactions than traditional serial experimentation (changing a single variable, one step at a time) and is more sparing in terms of the number of batches. A significant advantage of continuous over traditional batch processing is that greater numbers of experimental variants can be evaluated, especially with automation/PAT.
Unexpected significant interactions should be investigated. If they cannot be eliminated or reduced by formulation or process adjustments, a CMA may be required to control impact.
If level of incorporation of a basic excipient into a formulation is close to the minimum effective level, then the risk from variability for that excipient is greater (see percolation above). Ranging experiments to determine concentration sensitivity are useful. If basic excipient levels can be substantially reduced without impacting product performance, there is lower probability of excipient variability impacting product performance.
During the product life cycle, if variability in a basic excipient attribute unexpectedly starts to adversely impact finished product CQAs, then a new CMA may be required. The advantage of adding a CMA is that no prior regulatory approval is required. In the longer term (the next window of regulatory opportunity), a variation can be filed containing corrective formulation and/or process options, together with justification for retiring the now redundant additional CMA.
This article has been adapted from the CPhI 2021 New Year Report: Post Pandemic Trends & Excipient QbD.
References:
- IPEC Fédération asbl Rue Marie de Bourgogne 52 - 1000 Brussels – Belgium
T: +32 (0)2 213 74 40 - M: info@ipec-federation.org - Kano, N., Seraku, N., Takahashi, F. And Tsuji, S. (April 1984) Attractive quality and must-be quality, Journal of the Japanese Society for Quality Control (14-2), pp. 39-48.
- Carlin B, Wilson C, Excipients: Kano Analysis and Quality by Design, in Pharmaceutical Formulation: The Science and Technology of Dosage Forms ed G Tovey, Royal Society of Chemistry, 2018.
- O'Keeffe D, Campbell C, O’Donnell K. A Spectrum of Importance—Challenging the Concept of Critical/Non-Critical in Qualification and Validation Activities J Validation Tech Feb 29 2016
https://www.ivtnetwork.com/article/spectrum-importance%E2%80%94challenging-concept-criticalnon-critical-qualification-and-validation-ac