
Here's How USP mAb Standards Support Fast-Evolving Platform Approaches
By Niomi Peckham, director, biologics pipeline development, Science-Global Biologics, United States Pharmacopeia

The therapeutic potential of monoclonal antibodies (mAbs) came to the fore in the early 1970s when it was first demonstrated that pure protein could be obtained by fusing myeloma cells with the spleen cells of a mouse immunized with the desired antigen.1 In 1986, the FDA approved the first therapeutic mAb, muromonab or OKT3, for use in solid organ transplant recipients who had become steroid resistant; it took another eight years to approve the second mAb therapy. In the years since, significant advances in manufacturing have freed mAb production from previous limitations, allowing manufacturers to produce vast quantities of mAbs with increasing efficiency and at a lower cost. By 2019, nine mAb therapies were already among the 20 top-selling therapeutics, and in 2020, AbbVie's Humira (adalimumab) and Merck's Keytruda (pembrolizumab) generated $19.8 billion and $14.4 billion in revenues, respectively.1 Given the current demand for these products, it is not surprising that the number of mAbs in development and clinical trials is rapidly increasing.
Significant technological advances have made the discovery and development of mAb therapies more rapid and efficient. Recombinant DNA technology opened the possibility of not only custom designing mAbs but of producing them in a variety of host cell lines more amenable to commercial scale. Specifically, the development and optimization of mammalian cell lines, such as Chinese hamster ovary (CHO) cells, have revolutionized mAb production because manufacturers can now produce properly folded and modified proteins at levels that make the necessary up-front investments cost-effective. This technology has also increased the speed with which manufacturers can develop, scale up, and commercialize mAbs.
As the global biologics market grows, speed to market is a critical component of any development plan. This is especially true for mAbs, where the market grows more competitive every year. For this reason, first-to-market launches are highly valuable because they help the manufacturer gain and control market share. Analysis of nearly 500 drug launches between 1986 and 2012 shows that first-in-class players, on average, have a 6% market-share advantage over later entrants.2 Also, getting to market earlier gives the first-to-market therapy a longer lead time to establish a standard of care and extends the long-term profitability of the product.
The ability to make mAbs quickly is linked to the many types of modern production methods and the emergence of platform approaches. Today product developers have two primary options for scaling production. A company can build in-house capacity for mAb production or partner with a contract manufacturing organization (CMO) that has already invested in necessary facilities, equipment, and personnel. In-house manufacturing offers direct control over the process, but the time, labor, and capital expenditures required to implement this strategy can be cost-prohibitive for smaller developers. Alternatively, working with an experienced CMO is a viable strategy for companies that lack the resources and expertise to scale and operate the needed infrastructure. In general, an experienced CMO can make better use of valuable internal resources while building and maintaining a labor force with the necessary industry experience to mitigate risks that create barriers to getting to market quickly.
Standards For mAb Manufacturing
Developing and manufacturing mAbs requires comprehensive physicochemical and biophysical characterization. The analysis required is challenging, even for the most experienced manufacturers and CMOs, due to the complex nature of mAbs. Like other protein therapeutics, mAbs are post-translationally modified, which creates heterogeneity that must be monitored and controlled. Development and validation of the manufacturing process go hand-in-hand with analytical methods and standards that can measure critical quality attributes of the product with high confidence. These measurements are essential for determining if the manufacturing process is in control and delivering the desired target profile.
There are compendial standards for some biologics, but not for most mAb products. In these cases, standards-setting organizations can develop industry-accepted reference materials that may be used when product-specific standards are unavailable. The United States Pharmacopeia (USP) is one such organization, and as such, it spends significant resources on developing best practices and standards for the current and next generation of therapies. USP has worked for over a decade to create a clearly defined set of quality expectations for recombinant therapeutic mAbs. The first product of this effort was USP-NF General Chapter <129> Analytical Procedures for Recombinant Therapeutic Monoclonal Antibodies, which provides analytical procedures for testing common quality attributes of mAbs. 3 This chapter includes validated methods and system suitability criteria for purity assessments by chromatographic separation of size variants by liquid chromatography (SEC-HPLC), capillary sodium dodecyl sulfate electrophoresis (CE–SDS), and procedures for analysis of oligosaccharides and sialic acid in mAbs.
Next, USP focused on developing reference standards for mAbs that would be amenable to a "platform approach," both in terms of manufacturing and analytical development. The result was a set of mAb reference standards, which are recombinant humanized IgG1 expressed in CHO cells and purified using industry-standard production methods (USP mAb 001 RS, Monoclonal IgG1 Catalog #1445539, USP mAb 002 RS, Monoclonal IgG1 Catalog #1445547, and USP mAb 003 RS, Monoclonal IgG1 Catalog #1445595). Using USP developed best practices, these standards were rigorously tested and evaluated during multi-laboratory studies.
For example, because glycosylation is a critical quality attribute for lot release and characterization of mAbs, both before and after commercialization, the glycan heterogeneity of the reference standards was characterized by MS at both the intact level and after release using methods described in USP General Chapters <210> Monosaccharide Analysis and <212> Oligosaccharide Analysis. 4,5 Furthermore, the isoelectric point (pI) of the three mAbs was determined by capillary isoelectric focusing (cIEF) and imaged capillary isoelectric focusing (icIEF), and theoretical masses were confirmed by intact mass analysis performed using LC-high resolution mass spectrometry (HRMS) methods.
Table 1: USP mAb standards pI and molecular weight
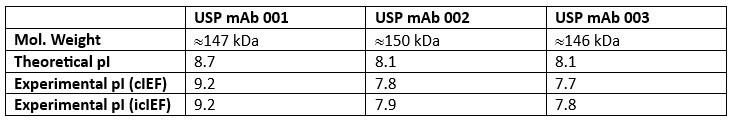
The isoelectric point (pI) values of the three mAb were determined by capillary isoelectric focusing (cIEF) and imaged capillary isoelectric focusing (icIEF). mAb 002 and mAb 003 have a lower pI relative to mAb 001 and each mAb shows a unique profile of acidic and basic variants. More analytical details and results can be found in the USP technical application note for mAb charge variant analysis.
The extensive characterization of the three mAb reference standards allows users to select the mAb that best matches the key attributes they need to assess. These standards are also ideal for CMOs that do not make their own products but still need appropriate reference materials for performance monitoring and training. For example, a large CMO based in South Korea recently shared their experiences using USP mAb reference standards at their facility, which manages 50 to 60 production projects annually. Depending on the project's specific demands, one mAb could be assessed by over 30 different assays and in-process control testing. Given this contractor’s production level, that testing panel could result in 4,000 assays supporting process validation runs for multiple projects over a year, which is nearly impossible without widely applicable standardized and validated platform methods.
This platform approach to analytical methods for process monitoring and evaluation of products requires a constant supply of reference standards. A commitment to update and maintain its standards inventory is one reason why CMOs use USP's mAb reference standards. In particular, one of our CMO customers needed an appropriately charged standard that was fully characterized to validate their icIEF assay for characterizing the pI of their mAb. Only USP's reference standards met the criteria for their platform needs because they were designed to standardize physicochemical testing, such as intact mass, charge heterogeneity, size variants, purity, and glycan analyses across multiple laboratories. In addition, they can also be used for a broad range of other applications, including as an internal assay control, reference material for method development (including platform technologies), and proficiency controls for method transfer and staff training.
Conclusions
Manufacturers have learned much about producing these complex therapeutics efficiently and consistently since the first mAb approval in the mid-1980s, but the learning process never stops. Advances in discovery are paving the way for new mAb scaffolds, such as fabs and bispecific antibodies. These new technologies, combined with constant cell line optimization, adoption of biologic continuous manufacturing methods, in-line/at-line monitoring, real-time release, and many more, are pushing the industry toward greater efficiency and productivity. But these advances don't come without risks and greater expectations for quality.
To succeed in mAb manufacturing, each stakeholder must leverage existing knowledge while staying flexible enough to adapt to future technologies. This must be accomplished quickly and under ever more stringent demands for demonstrating quality and control. For a CMO, staying flexible means developing platform analytical methods to handle many customer needs. But maintaining those assays within operational parameters requires reference standards. Manufacturers can choose to devote precious resources to developing their reference materials or rely on a trusted organization to create and thoroughly characterize those materials for them. If they choose the latter, then USP's mAb reference standards provide manufacturers with the best fit-for-purpose options for their specific analytical needs. Finally, USP is working on multiple additional IgG isotypes and impurities. Please contact the author to learn more.
References
- Mullard, A. (2021). FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov, 20(7), 491-495. 10.1038/d41573-021-00079-7
- Cha, M., & Yu, F. (2014). Pharma's first-to-market advantage. McKinsey & Company. https://www.mckinsey.com/industries/life-sciences/our-insights/pharmas-first-to-market-advantage#/
- USP. (2023a). <129> Analytical Procedures for Recombinant Therapeutic Monoclonal Antibodies. In USP43-NF38. The United States Pharmacopeial Convention. 10.31003/USPNF_M6297_02_01
- USP. (2023b). <210> Monosaccharide Analysis. In USP43-NF38. The United States Pharmacopeial Convention. 10.31003/USPNF_M5878_07_01
- USP. (2023c). <212> Oligosaccharide Analysis. In USP43-NF38. The United States Pharmacopeial Convention. 10.31003/USPNF_M5879_03_01
About the Author:
 Niomi Peckham is the director of pipeline development in USP Global Biologics. The pipeline development team works with scientific experts and stakeholders to develop physical and documentary standards used to support development and quality assessment of biopharmaceuticals. She holds a Master of Science in molecular and cellular biology from SUNY Stony Brook. She has spent most of her career at Pfizer and Alexion Pharmaceuticals, focusing on analytical development of protein-based biopharmaceuticals.
Niomi Peckham is the director of pipeline development in USP Global Biologics. The pipeline development team works with scientific experts and stakeholders to develop physical and documentary standards used to support development and quality assessment of biopharmaceuticals. She holds a Master of Science in molecular and cellular biology from SUNY Stony Brook. She has spent most of her career at Pfizer and Alexion Pharmaceuticals, focusing on analytical development of protein-based biopharmaceuticals.