
Factory Acceptance Testing, Site Acceptance Testing & Commissioning Activities, Oh My!
By Ajay Pazhayattil, Praveen Joseph, and Ciona Forsythe

Execution of factory acceptance testing (FAT) and site acceptance testing (SAT) is a business-critical activity when it comes to commissioning of a new system or equipment. The International Society for Pharmaceutical Engineering (ISPE) defines FAT and SAT as the inspection and static and/or dynamic testing of manufacturing systems performed to approve the systems for delivery and to support qualification of the systems.1 FAT is conducted at the equipment manufacturer’s site and SAT at the final site of installation.
FAT Is Key To Minimizing Downstream Issues
New biopharmaceutical equipment is customized and designed to fit the manufacturing operations model and product types being produced. The complex nature of the systems, the technology, and the intricacies associated with meeting business and regulatory expectations present constant challenges during the commissioning phase. Eliminating issues and errors up front allows you to minimize the number of business compromises that need to be made downstream. Unique requirements, such as accommodating the equipment’s dimensions for existing site drainage, piping, or room dimensions, need to be identified and addressed prior to shipment. In addition, new issues, such as position of the handle for accessibility, etc., can be identified when FAT is performed. The costs of addressing such issues, whether simple or complex, grow once shipped from the equipment manufacturing site. Given the intricacies of advanced technology, FAT is also the right place to verify software design specification, interface, and device integration.
A thorough assessment of the new unit post design and fabrication certainly reduces commissioning issues, which adds a definitive business value. However, to effectively measure the return on investment, businesses should entrust the same team with all commissioning activities, i.e., FAT, SAT, and commissioning. The metric should consider the number of issues observed post FAT in relation to the man-hours invested in the FAT and SAT activities. Addressing the issues at the equipment manufacturer’s site has a plethora of advantages, such as ease of sourcing components, quality of equipment/instrument used to test and correct, cost, etc. A FAT includes checks for safe access, interlocks, flow chart machine functions, orientation of door opening, equipment dimension, utility fittings, piping lengths, closeness, sensors, testing tamper resistance/seals, electrical requirements, and documentation such as hardware design specifications, drawings, and certificates. These aspects primarily relate to functionality, safety, fitness to user requirements, compliance of the system to GMPs, and data integrity. It is common to find inconsistencies post fabrication since there are possibilities of misses during translation of all the user requirement specifications (URS) and purchase order commitments into design/fabrication. An example is the use of a typical coated surface instead of a stainless-steel finish for environmental chambers. The product impact could be minimal since it is strictly used for storage of the unit’s post-secondary packaging. However, there could be an impact on the cleaning procedure that might be applied on-site. Making such changes at the installation site is impractical; hence, FAT is a must-do step versus nice to do, for critical systems.
FAT provides the opportunity for the equipment owner’s representatives to meet the equipment manufacturer’s expert teams, and the knowledge shared during the FAT sessions is critical to the long-term success of the commissioning process, i.e., through training. Identifying and involving the FAT team thus becomes critical. The test runs during FAT also provide first-hand experience observing the unit running. Identifying and resolving the punch list items during performance of the FAT allows for authorizing shipment of the equipment to site. This is a critical milestone prior to SAT; hence, it should only be completed by an authorized, trained, and approved subject matter expert.
Figure 1: FAT and SAT
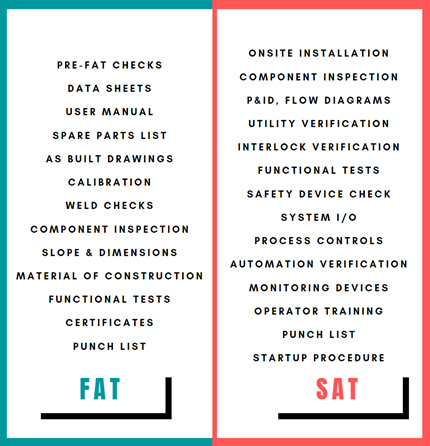
Upon receipt at the site and successful installation verification, critical aspects of the FAT are typically verified in the site environment through a SAT, including the interfaces and monitoring devices [Figure 1]. Utility capacity, automation, interface, and control issues/gaps usually are highlighted during this stage. Therefore, additional test cases to verify the potential failure modes need to be integrated into the SAT protocol.
Use A Responsibility Matrix For Optimal Efficiency
The off-site FAT, on-site SAT, and commissioning activities need careful planning for flawless execution. The commissioning2 and qualification plan (CQV plan) needs to be adjusted based on the extent of reliance on third parties for equipment expertise, test execution, commissioning project management, regulatory expertise, quality, automation, lab testing, etc. Table 1 provides guidance on the checks and balances required when utilizing external services for commissioning. A responsibility matrix provides clarity and ensures ownership of activities, along with optimal utilization of internal and external subject matter experts.
Table 1: Example - Balanced Responsibility Matrix
|
FAT/SAT Activity |
Execution Responsibility Matrix |
||
|
Equipment Manufacturer (Vendor) |
Approved External SME (Contractor) |
Customer Organization |
|
|
Scoping, identifying codes and regulatory references |
Provide supporting evidence |
Draft and/or review, provide SME inputs. |
Drafted and/or approved by regulatory compliance |
|
User requirement specification (URS) |
Provide consultation. |
Draft and/or review, provide SME inputs. |
Drafted and/or approved by user department |
|
Data sheet |
Provide per client URS requirement.
|
Review, provide SME inputs for revisions where needed. |
Review and approval by internal engineering and stakeholder departments. |
|
Drawings |
Provide per client design requirements. |
Review support. |
Review and approval by internal engineering. |
|
FAT inspection/test plan/protocol |
Draft and review. |
Collaborate with vendor to finalize the plan. Provide SME inputs. |
Collaborate, review, and approve by internal engineering and stakeholder departments. |
|
Operating Procedures |
Provide operating manual. |
Draft and/or provide SME inputs. |
Internal user department and stakeholders draft and/or approve. |
|
Calibration records |
Perform calibration and provide records. |
Perform on-site calibration where required and provide records. |
Perform on-site calibration and/or approve calibration reports. |
|
Calibration schedule |
NA |
Provide calibration frequency justification. |
Develop calibration schedule and maintain. |
|
Certificates, material of construction, finish etc. |
Provide certificates |
Verify and provide SME input. |
Verification and acceptance. |
|
Raw data review |
Vendor’s internal QA. |
On-site verification support. |
Reviewers and QA. |
|
Participation |
Vendor PM, technologists, SME |
On-site or virtual execution support. |
Engineering, qualification, and user departments. |
|
Inspection: Ex. weld, name plate, drain, dimensions, documents |
Vendor technologists, SME. |
On-site or virtual execution support. |
Engineering, qualification, and user departments. |
|
Tests: Ex. pressure, vacuum, component functionality, controls. |
Vendor technologists, SME. |
On-site or virtual execution support. |
Engineering, qualification, and user departments. |
|
Data integrity review |
Vendor QA. |
On-site verification. |
|
|
Punch list resolution |
Vendor PM, technologists, SME. |
Verify and provide SME input. |
Internal user department, engineering, and stakeholder approval. |
Defining The Goals In A CQV Plan & Leveraging Checkpoints
In addition to the responsibility matrix (including responsibilities of approved contractors), a CQV plan must have other critical components for transparency throughout the project and for robustness. The CQV plan should define the approach for the URS, system impact assessment (SIA), design specification (DS), hardware/software specification (HDS/SDS), functional risk assessment (FRA), FAT, SAT, commissioning, and qualification (IQ/OQ/PQ) documentation, traceability matrix, and stage gate requirements for each equipment. In addition, the approach to deviation and change management during commissioning and qualification must be defined in the plan. The site engineering team should be responsible for commissioning and validation of the qualification activities. They can rely on project managers and consultants to drive the project and to provide status updates. Where required, the sampling plan needs to be statistically justified by a trained statistician, as stipulated by guidance.3 Different approaches can be taken when qualifying direct and indirect impact systems. It must be verified that all test methods/procedures used are qualified prior to use.
To apply a risk-based approach,4 there needs to be documented justification at each stage that would withstand any regulatory scrutiny. It has been found that 45% of the data integrity issues in warning letters were associated with operations related data.5 New production equipment, with a substantial amount of automation, monitoring, parameters, and attributes, generates critical data. Data governance, control, and assurance of traceability and integrity need to be considered during early commissioning. The controls become even more critical when risk-based leveraging is applied. Though the CQV plan can potentially identify leveraging opportunities, the outcome of the test cases and the integrity assurance of the vendor-provided data need to be considered prior to officially accepting the test to be leveraged. Adequate stage gate documented decision steps need to be integrated into a concise CQV plan.
Conclusion
Developing your FAT and SAT plans, executing them, and maintaining integrity throughout the process requires careful, meticulous planning and continual re-evaluation of the plan. This is even more critical during the pandemic, when teams work across the globe virtually and activities are fast tracked. The development of a fit-for-purpose CQV plan with a clear definition of vendor and contractor roles and responsibilities,6 continual reconfirmation against the plan after every critical execution, providing justification of any changes to the plan, and incorporating enough stage gate points for the assessment are key aspects to be considered. The goal is to assure traceability and maintain integrity throughout the project, where multiple external parties are involved, both virtually and on-site. Where enough supporting evidence is not available, the strategy on leveraging needs to be reconsidered. It is also ideal to have the team goals and responsibilities established. Maintaining the team until the CQV planned activities are complete is critical since much knowledge may remain tacit in a fast-paced commissioning scenario. Adequate mechanisms need to be in place within the CQV plan to document and convert the tacit knowledge into internal explicit knowledge at the earliest opportunity. Applying the insights discussed in this article can add value and greatly reduce potential downstream issues during a critical CQV project.
References
- ISPE (2019). Baseline Guidance, Commissioning and Qualification, 2nd ed., International Society for Pharmaceutical Engineering.
- Blackburn, T. (2004). A Practical Approach to Commissioning and Qualification-A Symbiotic Relationship, Pharmaceutical Engineering.
- FDA (2011). Process Validation: General Principles and Practices, FDA. https://www.fda.gov/files/drugs/published/Process-Validation--General-Principles-and-Practices.pd
- Pazhayattil, A. (2021). Biopharma Equipment Design, Qualification and Monitoring Model, Journal of Validation Technology.
- PDA (2020). Technical Report No. 84: Integrating Data Integrity Requirements into Manufacturing & Packaging Operations, Parenteral Drug Association.
- CFR Part 211, Subpart B Section 211.34 Consultants.
 About The Authors:
About The Authors:
Ajay Pazhayattil is a pharmaceutical management consultant leading technical operations, quality assurance, and regulatory compliance risk mitigation/remediation projects. He has conceived and implemented novel methodologies applying sound pharmaceutical science principles. He is an industrial pharmacist with experience in solid dose, liquid, and parenteral dosage forms.

Praveen Joseph is a quality engineer with more than 20 years of experience managing various projects across product life cycles within the biopharmaceutical industry. His expertise is in R&D, technology transfer, validation, quality, systems, and compliance. He is a thought leader in technical operations, CQV, cold chain, and compliance.
 Ciona Forsythe’s experience in the pharmaceutical and biologics (cell and gene therapy) industry includes validation, aseptic processing, sterilization, production, technology transfer, engineering, quality operations, regulatory compliance, process management, process validation, and consulting. She is skilled in strategic risk identification and mitigation.
Ciona Forsythe’s experience in the pharmaceutical and biologics (cell and gene therapy) industry includes validation, aseptic processing, sterilization, production, technology transfer, engineering, quality operations, regulatory compliance, process management, process validation, and consulting. She is skilled in strategic risk identification and mitigation.