
EMA's New Guideline For Synthetic Peptides: A More Explicit CMC Playbook For A Growing Therapeutic Class
By Tim Sandle, Ph.D.

The European Medicines Agency’s Guideline on the Development and Manufacture of Synthetic Peptides, adopted in December 2025 and effective from June 1, 2026,1 is an important regulatory development for peptide manufacturers. It addresses a long-recognized gap: synthetic peptides do not fit neatly within the traditional regulatory frameworks for small molecules or biologics, yet they present quality, manufacturing, and analytical challenges that require more specificity than either framework has historically offered.
The EMA guideline is best understood as a peptide-specific chemistry, manufacturing, and controls (CMC) bridge document — one that translates general ICH and EU dossier principles into practical expectations for solid-phase peptide synthesis (SPPS), fragment condensation, conjugation, impurity characterization, and comparability.
Peptides: The Regulatory Middle Ground
At its core, the document acknowledges the scientific reality that peptides occupy a regulatory middle ground. The EMA states explicitly that synthetic peptides lie “at the interface of small molecules and proteins” and notes that important parts of the ICH framework — such as ICH Q3A/B, Q6A/B, and ICH M7 — do not fully cover peptide-specific issues. The result is a guideline that is less a radical departure than a structured supplement: it retains the Common Technical Document (CTD) organizational format, and it leans on established standards such as ICH Q7, Q9, Q12, Q13, and Q14. However, the guideline also adds tailored expectations where peptide complexity makes general principles insufficient.
One of the strongest aspects of the guideline is the level of operational detail expected for manufacturing descriptions. For SPPS processes (which manufacturers use to build peptides step-by-step on a solid support [resin]),2 applicants are expected to clearly describe repeated cycles of deprotection, washing, coupling, and capping, without necessarily repeating identical text for every cycle, provided the process is still transparent and traceable. However, the final cleavage and deprotection steps must be described in detail, including scavengers and critical reagents.
EMA also brings clarity to industrial realities such as batch splitting, sub-batch pooling, routine repurification of side fractions, and repeated coupling steps. These practices are acceptable, but only if they are prospectively defined, justified, traceable, and embedded within the validated commercial process rather than presented as ad hoc corrective actions. This is important because many peptide dossiers historically suffered from under-described purification and pooling logic, even though those operations can materially shape the final impurity profile.
The guideline is equally notable for how it treats starting materials. EMA applies ICH Q11 logic rigorously:3 applicants must justify the designation of peptide starting materials and explain the impact of their impurity profiles on the final active substance. Protected amino acid derivatives are acceptable starting materials, and some dipeptides or short fragments may also qualify if scientifically justified. However, longer peptide fragments are usually viewed more cautiously and may be considered intermediates rather than starting materials unless a convincing case is made. Preloaded resins containing the first amino acid may qualify as starting materials, whereas unloaded resin itself does not. This is a risk-based position; however, this approach will push manufacturers toward earlier and more extensive scrutiny of supplier controls, stereochemical integrity, and impurity carryover at the building block level.
With characterization expectations, the EMA expects confirmation of primary structure and, where relevant, higher-order structure using suitable orthogonal analytical techniques. Mass spectrometry, amino acid analysis, NMR, circular dichroism, peptide mapping, and, in some cases, biological assays all appear in the recommended characterization toolbox. This is scientifically appropriate. Although many peptides do not have meaningful or persistent higher-order structure, some do display relevant conformational features, oligomeric states, or aggregation tendencies that may affect biological performance or comparability.4
The guideline, therefore, avoids a one-size-fits-all model: if a sponsor can show that higher-order structure is absent or not relevant, the burden can be reduced; if not, deeper physicochemical and, sometimes, functional characterization are expected.
The most consequential part of the guideline for many developers will be impurity control. EMA emphasizes that purity is one of the most important critical quality attributes for synthetic peptides and offers a detailed taxonomy of peptide-related impurities: stereoisomers, deletion sequences, truncated sequences, insertion sequences, cleavage-related by-products, degradation products, and high molecular weight impurities such as dimers, oligomers, and aggregates.
It also emphasizes non-peptide impurities, including reagents, solvents, elemental impurities, and potentially mutagenic impurities. Importantly, the document repeatedly highlights the risk of co-eluting impurities and therefore expects highly specific analytical methods, sometimes more than one, with sensitivity sufficient to support a reporting threshold of 0.1%. That expectation reflects the real analytical difficulty of peptide control: impurities are often structurally close to the target, not easily resolved, and not necessarily well served by a single chromatographic method.
The document also provides practical clarity on specification-setting. Since peptides are excluded from ICH Q3A, the usual small molecule impurity framework does not apply directly. Instead, the guideline points to the European Pharmacopoeia general monograph Substances for Pharmaceutical Use,5 under which peptide-related impurities should be reported above 0.1%, identified above 0.5%, and qualified above 1.0%. This is significant because it creates a more explicit benchmark for peptide dossiers than many sponsors have worked with in the past. At the same time, the document sensibly warns that acceptance criteria cannot simply be copied from formal thresholds; they must also be grounded in batch history, process understanding, and the impurities actually qualified in nonclinical and clinical material. This is a mature regulatory stance, but it is also one that will demand better life cycle impurity knowledge from industry.
A key section deals with medicinal product considerations, especially for parenteral peptide products. The guideline states that terminal sterilization should remain the preferred option unless the sponsor can show it is unsuitable, and it explicitly says that exceeding impurity qualification thresholds is not, by itself, sufficient reason to reject terminal sterilization. Instead, sponsors are expected to optimize formulation, container closure, and process design to create the best possible sterilization strategy and, where degradation occurs, to qualify those degradants appropriately. This is a demanding position, but also a pragmatic one. It aligns peptide development with broader sterile product principles and discourages routine reliance on aseptic processing without a robust technical justification.
The comparison section for synthetic peptides developed against a biological reference product is especially interesting because it lays out an EU view that is adjacent to, but distinct from, the biosimilar paradigm. EMA states clearly that chemically synthesized peptides cannot follow the biosimilar route because they are not biological substances. Nevertheless, when a synthetic peptide references a biological product, the agency expects the sponsor to follow the principles of biosimilarity.
Implications For Industry
In terms of the implications for manufacturers:
- Peptide development programs will need stronger analytical platforms earlier in development, especially orthogonal methods capable of resolving closely related impurities and detecting high molecular weight species.
- Control strategies will increasingly start upstream, at the level of amino acid derivatives, loaded resins, linkers, and conjugation components, rather than being deferred to final API release testing.
- Sponsors should expect greater scrutiny of process narratives, purification logic, repurification decisions, and comparability when multiple manufacturing routes or suppliers are used.
- The document raises the bar for life cycle consistency: changes in process, methods, or starting material sourcing will need to be supported by clearer scientific rationale and, where needed, comparative data packages.
The guideline should reduce regulatory ambiguity, but it also increases expectations for process knowledge and dossier maturity.
Comparison With The U.S. FDA
Compared with the FDA, the EMA’s approach is broader from a CMC perspective (refer to Table 1 for a comparative summary). The FDA does not currently have a directly equivalent overarching chemistry, manufacturing, and control guidance for synthetic peptides spanning innovator, life cycle, and finished product issues. The most relevant FDA document is the 2021 guidance ANDAs for Certain Highly Purified Synthetic Peptide Drug Products That Refer to Listed Drugs of rDNA Origin,6 which is narrower in scope and focused on when a synthetic peptide that references a recombinant peptide may be submitted via the ANDA pathway rather than an NDA. That FDA guidance emphasizes active ingredient sameness, comparative impurity studies, and immunogenicity-related impurity evaluation for specific follow-on generic contexts, rather than providing a full peptide manufacturing framework.
That difference in scope matters. EMA’s new guideline speaks to the complete product quality package: manufacturing process design, control of materials, characterization, specifications, conjugation, finished product development, sterilization, stability, and investigational submissions. By contrast, the FDA’s peptide-specific guidance is more narrowly concerned with equivalence and impurity risk for certain generic synthetic peptides that reference rDNA-origin listed drugs.
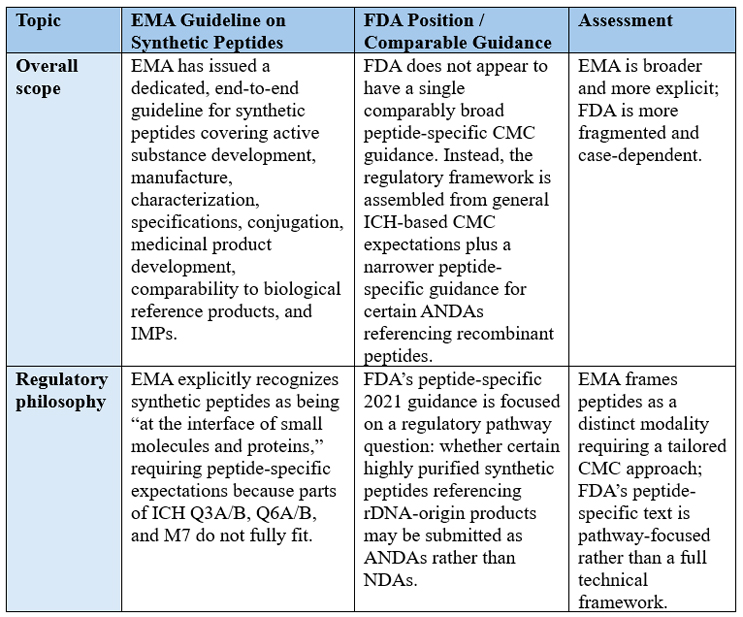
Table 1: Comparison between EMA and FDA approaches for synthetic peptides
There are areas of convergence, as Table 1 indicates. Both agencies place major weight on impurity control, orthogonal analytics, and the possibility that peptide-related impurities or aggregates could alter safety, efficacy, or immunogenicity. Both also recognize that advanced analytical technologies now allow a level of structural and impurity characterization that was not feasible years ago. FDA’s 2021 guidance similarly stresses comparative impurity profiles and the need to justify new or higher-level impurities, while EMA extends that same logic across the broader development life cycle.
New Regulatory Thinking
Overall, the EMA guideline is a timely and constructive document. Its main strength is not that it invents entirely new standards but that it consolidates peptide-specific regulatory thinking into a coherent and practical framework. For manufacturers, this should improve predictability.
References:
- EMA. Guideline on the Development and Manufacture of Synthetic Peptides, European Medicines Agency, EMA/CHMP/CVMP/QWP/367182/2025: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-development-manufacture-synthetic-peptides_en.pdf
- Zero J, Tyler TJ, Cronin L. Universal peptide synthesis via solid-phase methods fused with chemputation. Nat Commun. 2025;16(1):7322. doi: 10.1038/s41467-025-62344-2
- ICH. Development And Manufacture Of Drug Substances (Chemical Entities And Biotechnological/Biological Entities) Q11, 2012, International Conference On Harmonization Of Technical Requirements For Registration Of Pharmaceuticals For Human Use: https://database.ich.org/sites/default/files/Q11_Guideline.pdf
- Parra Bravo C, Naguib SA, Gan L. Cellular and pathological functions of tau. Nat Rev Mol Cell Biol. 2024 Nov; 25(11):845-864. doi: 10.1038/s41580-024-00753-9
- General monograph “Substances for pharmaceutical use,” Monograph Nr 2034, European Pharmacopoeia, Edition 12, 2025
- FDA. ANDAs for Certain Highly Purified Synthetic Peptide Drug Products That Refer to Listed Drugs of rDNA Origin, Guidance for Industry, 2021: https://www.fda.gov/media/107622/download
About The Author:
 Tim Sandle, Ph.D., is a pharmaceutical professional with wide experience in microbiology and quality assurance. He is the author of more than 30 books relating to pharmaceuticals, healthcare, and life sciences, as well as over 170 peer-reviewed papers and some 500 technical articles. Sandle has presented at over 200 events and he currently works at Bio Products Laboratory Ltd. (BPL), and he is a visiting professor at the University of Manchester and University College London, as well as a consultant to the pharmaceutical industry. Visit his microbiology website at https://www.pharmamicroresources.com.
Tim Sandle, Ph.D., is a pharmaceutical professional with wide experience in microbiology and quality assurance. He is the author of more than 30 books relating to pharmaceuticals, healthcare, and life sciences, as well as over 170 peer-reviewed papers and some 500 technical articles. Sandle has presented at over 200 events and he currently works at Bio Products Laboratory Ltd. (BPL), and he is a visiting professor at the University of Manchester and University College London, as well as a consultant to the pharmaceutical industry. Visit his microbiology website at https://www.pharmamicroresources.com.