
Building AAV Manufacturing Capacity For Large Patient Diseases
By Antony Hitchcock, principal and owner, AGH Bioconsulting

Over the last decade, we have witnessed the development of multiple AAV-based gene therapies offering transformational cures to patients with debilitating and often fatal conditions. We are now seeing some of these products receiving regulatory approval (Table 1).1 However, the exceedingly high prices levied on these products have limited adoption as healthcare providers are unable to justify or afford these very high upfront costs compared to existing therapies, especially for indications with large patient groups such as hemophilia. The UK National Institute for Health and Care Excellence’s refusal to recommend products such as Hemgenix treatment for hemophilia B in August this year2 in place of existing replacement therapy exemplifies this. A key factor behind these high prices is that the majority of products largely have been developed for rare or ultra-rare conditions with limited markets, this means that in addition to the very high production costs of these products, developers are seeking to recover development costs from a small number of patients.
A key question going forward is this: could AAV-based gene therapies be used to treat larger patient populations such as cardiovascular and neurological conditions, where development costs may be spread over more patients making products more affordable and adoptable by healthcare providers?
To do this, there are key issues that must be addressed with regard to product safety and improvements in manufacturing processes.
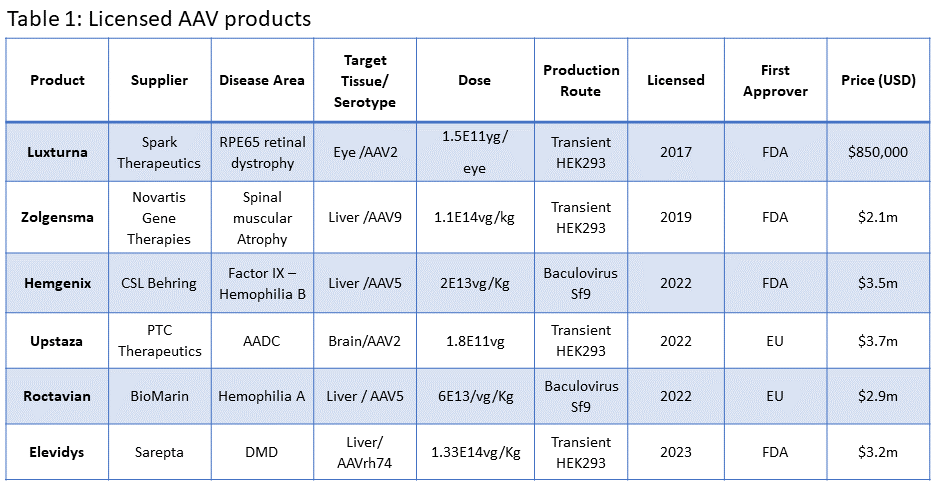
To better understand the manufacturing challenge of treating large patient groups, it is useful to look at potential manufacturing demands of some of the current, licensed products based on current productivity levels and largest production scales (Table 2). We can see that current production processes are only able to produce a few hundred doses per batch and for high-dose therapies as few as 10 per batch. Without significant improvements in dosing and manufacturing, it is not going to be possible to treat large patient groups.
Addressing The Cost Issues
While it is possible to focus solely on improvements in production systems to address manufacturing shortfalls, arguably, we need to take a more holistic view and assess what can be done to reduce dose levels to mitigate manufacturing demands. When we look at commonly used systemic approaches, in some cases we are treating patients with ca 200 vectors per cell in the body. This is significantly elevated for specific organs such as the liver and has been linked to undesired clinical side effects.
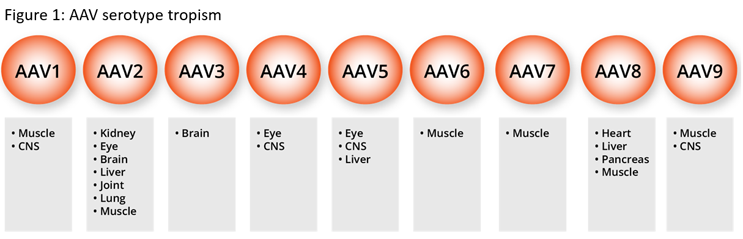
AAV vectors have been used as gene delivery vehicles as they offer a level of tissue specificity (Figure 1); however, many of the commonly used vectors have broad tissue specificity. The need to increase vector specificity and reduce immunogenicity through vector engineering has been recognized for a number of years. To this end, for over 20 years groups of scientists have looked at engineering vectors to improve safety and targeting with a range of techniques and increasingly using artificial intelligence-based approaches.3,4,5 Furthermore, we have seen similar approaches to the improvements in the design of the gene of interest (GOI), with regard to improvement to achieve greater levels of expression and the use of tissue-specific promotors to prevent off-targeting of vectors.
While these activities were historically in the domain of academic and internal research groups, we are now seeing several academic spin-outs offering vector engineering services, giving smaller development companies access to engineered vectors. In doing so, the use of engineered vectors is becoming increasingly mainstream.
There is now an increasing number of engineered vectors in clinical trials6 and the clinical data being generated from these studies indicate that the dose levels with engineered vectors may be as much as 2 logs lower than with native vectors. This significant reduction in dose levels has the potential to markedly reduce vector demands while addressing issues of patient safety including immunogenicity.
Manufacturing Developments
Let’s take a deeper dive into manufacturing issues and the unsolved problems of restricted supply and high production costs.
There are several potential approaches to the production of vectors for clinical use. The most widely-used approach is the triple transfection route in HEK293 cell lines. Vector genes are coded onto plasmids, which are then mixed with a transfection reagent and added to the cell culture systems. Initial transfection-based production processes were performed in adherent cell culture systems, which, in turn, restricted development and production scales because of the limited available production systems and development tools. As a consequence, alternative approaches, including the baculovirus systems which can be performed in scalable suspension platforms, were adopted by some product developers looking to meet clinical and predicted demands for diseases such as hemophilia with high product demands.
Baculovirus
In the baculovirus-mediated production in Sf9 insect cells, vector genes are usually coded into one or more baculovirus, and production occurs in the cells that contain their own helper functions. There is no singular approach to vector production in insect cells, and significant development has occurred within this platform to address issues around baculovirus stability and heterogeneity of capsids and high levels of empty capsids.7 Until recently these have been around five times more productive than the transient production routes, with reported productivities in the region of 1-4E11vg/ml. However, while there is limited published data, there is a perception that vectors produced in insect cells have a lower level of infectivity than those produced in human cell lines and hence higher doses may be required to treat patients potentially outweighing any improvement in productivity.8
Triple transfection
Transfection processes were initially based upon academic methodologies and reagents used for generating and adding plasmid DNA complexes to cells, however, there have been significant efforts in recent years in the industrialization of these processes to support the production of vectors for clinical studies and commercial supply.9 Initially, these developments moved developers away from adherent cultures to suspension culture systems and, while the suspension systems were initially of lower productivity, they offered a more readily-scaled production platform. This was because they offered access to predictive scale-down models and a vast amount of industrial experience with these technologies. It allowed for intensive development studies to be performed to identify critical process parameters, which has allowed for process optimization and intensification.
More recent developments have focused on transfection processes, in terms of amounts and ratios of the plasmids used, improvements to transfection reagents, and the procedures used to generate and add the plasmid DNA complexes to cell culture systems. Additionally, we have also seen significant improvements in cell culture systems with the adoption of clonal cell lines and the adoption of perfusion platforms to achieve increased cell densities.
These improvements in transfection procedures and cell culture have allowed for the challenges around process scale-up to be addressed with regard to consistency of titers and retention of product quality. We now see companies reporting robust production processes at the 2,000L scale achieving comparable titers to those seen at small scales. These are achieving titers in the region of 1E11vg/ml representing 2E17vg in a 2,000L bioreactor.
Going forward, we see companies reporting productivity levels in the region of 1E12vg/ml in transient cell culture systems at small and potentially larger scales through the use of novel transfection reagents.10 This represents a log increase on titers that are currently being used in production systems, and these may be further improved in high-density cell culture systems.
Stable cell lines
After many years of development, we see the emergence of stable cell lines as viable production platforms where the rep/cap, GOI, and helper functions are stably integrated into the production chromosome and require the control of small molecule inducers to initiate vector production.11 They offer a simplified production approach, avoiding the need for plasmid or viral seeds and should be readily scaled and optimised to maximise productivity. The generation of these cell lines has, to date, proven technically challenging; however, we now see companies offering the development of the cell lines. Early studies report productivity as high as 5E12vg/ml. In all, these changes represent up to a 1.5 log improvement on current titers.
Downstream processing
A final area of potential improvement in productivity is in downstream processing. The key challenges in the recovery and purification of AAV vectors have been low production titers and the requirement to separate empty and full capsids. Over recent years, we have seen a standardization of processes, the key variation being the approaches to remove empty capsids.
The critical areas for improving process yields have been the improvements in the initial recoveries during clarification steps, the avoidance of initial TFF operations, and, critically, improvements in the recoveries from the affinity capture steps. The separation of full and empty capsids is still challenging, but there are improvements in this area although they may be serotype-dependent. The impact of these developments is that we have seen process recoveries increase from less than 25% to reported recoveries of around 50%, representing a 0.5 log improvement.
Summary
Taking a holistic view of these developments, it is apparent that we not only are seeing significant potential reductions in required dose levels where it may be possible to reduce anticipated vector requirements by as much as 1.5 to 2 logs. We also see developments in manufacturing processes that could realistically result in a 1-to-1.5 log increase in production titers. If we update Table 2 with these numbers (Table 3), we can see that the changes are significant in that all current annual demand for licensed product could be potentially sourced from a single 2,000L batch. This represents a profound change with regard to the potential to produce tens of thousands to hundreds of thousands of doses from a single batch, making the treatment of large population groups a realistic possibility.
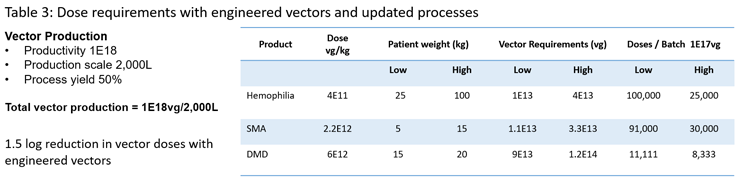
These developments have the potential to significantly impact the ability to supply material to clinical trials, which is currently problematic in terms of the need for large-scale processes to supply early-phase studies. In turn, it should also make a meaningful impact on the overall cost of goods and the supply for clinical studies. As it should be possible to produce large numbers of doses from a relatively small-scale production scale of less than 2,000L, additionally the use of stable cells should also allow for production scales of over 2,000L if needed.
Looking into the future, we have good reasons to feel optimistic about AAV therapies’ potential to transition from the domain of orphan conditions to more mainstream diseases and give more patients access to new and revolutionary medicines. These developments should also address the cost challenge and encourage investors to continue supporting life-changing medicines.
References
- https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products
- https://www.nice.org.uk/guidance/gid-ta10699/documents/draft-guidance
- Engineering adeno-associated virus vectors for gene therapy. Chengwen Li1 and R. Jude Samulski, Nature Reviews Genetics volume 21 | April 2020 | 255
- Fantastic AAV Gene Therapy Vectors and How to Find Them—Random Diversification, Rational Design and Machine Learning. Jonas Becker, Julia Fakhiri , Dirk Grimm. Pathogens 2022, 11(7), 56; https://doi.org/10.3390/pathogens11070756 Received: 23 May 2022 / Revised: 30 June 2022 / Accepted: 1 July 2022 / Published: 3 July 2022
- Engineering adeno-associated viral vectors to evade innate immune and inflammatory responses. Church et Al, Science translational medicine 10 Feb 2021Vol 13, Issue 580, https://doi.org/10.1126/scitranslmed.abd3438
- Au HKE, Isalan M and Mielcarek M, (2022) Gene Therapy Advances: A Meta-Analysis of AAV Usage in Clinical Settings. Front. Med. 8:809118. doi: 10.3389/fmed.2021.809118
- Joshi, Pranav R.H., Venereo-Sanchez, Alina, Chahal, Parminder S., Kamen, Amine A. Advancements in molecular design and bioprocessing of recombinant adeno-associated virus gene delivery vectors using the insect-cell baculovirus expression platform. Biotechnol. 04 Dec 2020 J. 16. 4 1860- 676, https://doi.org/10.1002/biot.202000021
- Rao, R.; Farraha, M.; Logan, G.J.; Igoor, S.; Kok, C.Y.; Chong, J.J.H.; Alexander, I.E.; Kizana, E., Performance of Cardiotropic rAAV Vectors Is Dependent on Production, Method, Viruses. 2022, 14, 1623. https://doi.org/10.3390/v14081623
- Packaging Across Multiple AAV Serotypes and Genomes. Tang Q, Keeler AM, Zhang S, Su Q, Lyu Z, Cheng Y, Gao G, Flotte TR, Molecular Therapy: Methods & Clinical Development (2023), doi: https://doi.org/10.1016/j.omtm.2023.05.004.
- A novel dual-plasmid platform provides scalable transfection yielding improved productivity and packaging across multiple AAV serotypes and genomes. Gilmore R, Stanvick M, Burnham B, Gagnon J, Iwuchukwu I, Yang G, Ghazi I, Meola A, Dickerson R, Thiers T, Mustich L, Hayes A, Rivas I, Lotterhand J, Avila N, McGivney J, Yin J, Kelly T Molecular Therapy: Methods & Clinical Development Vol. 29 June 2023
- Transfer and scale-up from 10 L BioBLU to Allegro STR 50 and STR 200 Bioreactors. David Mainwaring, Amar Joshi, Juliana Coronel & Christian Niehus, Cell & Gene Therapy Insights 2021; 7(9), 1347–1362 DOI: 10.18609/cgti.2021.178
About the Author:
 Tony Hitchcock is the principal and owner of AGH Bioconsulting. He has spent his decades-long career in the in the biotechnology field, with over 30 years in the production of complex biologic for clinical trials in the European Union and U.S. He has worked in areas of process development and manufacturing with experience in engineering and process systems. He has worked on the development of more than 30 products for clinical trials including plasmid DNA, viral and bacteriophage products, and recombinant proteins from microbial, mammalian, and insect cell sources. Contact him at tony@aghbioconsulting.com.
Tony Hitchcock is the principal and owner of AGH Bioconsulting. He has spent his decades-long career in the in the biotechnology field, with over 30 years in the production of complex biologic for clinical trials in the European Union and U.S. He has worked in areas of process development and manufacturing with experience in engineering and process systems. He has worked on the development of more than 30 products for clinical trials including plasmid DNA, viral and bacteriophage products, and recombinant proteins from microbial, mammalian, and insect cell sources. Contact him at tony@aghbioconsulting.com.