
Achieving Annex 1 Compliance In Sterile Manufacturing, Part 3: Building A Compliant Program
By Bikash Chatterjee, president and CSO of Pharmatech Associates, a USP Company and USP Microbiology

Parts 1 and 2 of this series identified the compliance gaps most consequential for Annex 1-compliant environmental monitoring programs and introduced the objective measurement tools, which include standardized recovery reference surfaces, validated inoculum delivery systems, and compendially anchored endotoxin reference standards that can address each gap.
What remains is embedding those tools within a program architecture designed to generate, interpret, and act on objective data systematically, across the full life cycle of the contamination control strategy (CCS). Part 3 describes what a well-constructed Annex 1-compliant program looks like at each stage, from gap assessment through governance, and how USP measurement tools integrate at each step. A compliance mapping table and conclusion follow.1,2
This is part 3 of a three-part series. Part 1 covered real-world compliance failures. Part 2 covered objective measurement tools.
What Good Looks Like Across The CCS Life Cycle
The measurement tools described in part 2 of this series address specific, documented gaps in EM program performance. But the value of objective data is only realized when it is embedded in a program architecture that is designed to generate, interpret, and act on it systematically. This section describes what a well-constructed Annex 1-compliant program looks like at each stage of the CCS life cycle, from initial gap assessment through ongoing governance, and how standardized reference materials integrate into each stage to provide the objective measurement foundation that can address regulatory expectations.
Start With An Honest Assessment: Gap Analysis As Scientific Baseline
The first step in building or rebuilding a compliant EM program is an objective assessment of where the current program stands relative to Annex 1’s expectations. A genuine gap analysis evaluates the scientific defensibility of the program’s design, the reliability of the data it generates, and the degree to which personnel performance contributes to or undermines data quality. A rigorous gap analysis should encompass three dimensions. The first is program design: whether sampling locations, frequencies, and methods are justified through a documented risk assessment linked to the CCS. The second is method characterization: whether recovery efficiency data is available for the contact plates and swabs in use, and whether that data is method-specific and site-specific rather than drawn from generic literature. The third is personnel performance: whether the facility can demonstrate, with objective data rather than training records, that analysts performing surface sampling are capable of recovering contamination at an acceptable and consistent rate. Deploying standardized recovery reference materials across shifts, rooms, and sampling teams during a gap assessment generates the baseline performance data that answers the third question and frequently reveals the most significant compliance exposure. Sites that have never measured recovery efficiency are often surprised by the degree of variability between analysts and by the proportion of operators whose routine technique falls below a defensible performance threshold. That data, however uncomfortable, is precisely what is needed to understand the true state of the EM program and prioritize remediation accordingly.3
Program Design: Building Scientific Justification Into Every Decision
An Annex 1-compliant EM program is built on science-based design decisions that are explicitly linked to the CCS and documented with scientific rationale. Sampling locations are justified through contamination risk assessment. Sampling frequencies reflect the risk profile of each location and historical performance data. Alert and action levels are derived from historical data and expressed in operationally meaningful, statistically defensible terms. Contact plate selection is a decision that requires particular attention. The multisite recovery study discussed in part 1 demonstrated substantial variation in recovery efficiency across 16 commercially available contact plate brands, meaning that the choice of contact plate is not a procurement decision but a scientific one. Recovery efficiency data generated using standardized reference surfaces allows facilities to compare plate performance under their own field conditions, with their own analysts and procedures, providing the site-specific justification that Annex 1 describes rather than relying on manufacturer claims.4
Media qualification must similarly be built on a reproducible, documented foundation. GPT performed with a standardized inoculum delivery system provides lot-to-lot consistency data that supports both the release of individual media lots and the long-term reliability of the media qualification program. When EM media performance is consistent and well documented, the interpretation of routine monitoring data becomes more straightforward, excursions are less likely to be attributed to media performance rather than environmental conditions.
Personnel Qualification: From Observation To Demonstrated Competency
A compliant personnel qualification program for surface sampling must do two things that observational approaches cannot: confirm that analysts can recover contamination from a surface reproducibly and at an acceptable rate and confirm that they can do so without introducing contamination in the process. These are distinct capabilities that require distinct assessment methods, and both must be documented in a format that supports trend analysis and periodic reassessment.5
Initial qualification should be conducted before analysts are authorized for routine monitoring, using standardized reference surfaces that provide a defined recovery challenge under conditions representative of actual sampling use. Pass/fail criteria should be established in advance, documented in the qualification protocol, and applied consistently across all personnel. Qualification records should capture individual recovery percentages, not just pass/fail outcomes, to enable trend analysis across the analyst population over time.
Periodic requalification at defined intervals maintains the integrity of the qualification program over time and provides the longitudinal performance data that supports CCS reviews and investigation activities. Requalification should also be triggered by specific events: following deviations attributable to sampling technique, after significant procedural changes, or when EM trend analysis identifies anomalies that may be method-related rather than environmental.

Trending And Investigations: Making The Data Work
Environmental monitoring data is only as useful as the analytical framework applied to it. A compliant trending program does not simply chart CFU counts over time, it integrates multiple data streams to build a picture of environmental performance that can distinguish signal from noise and support meaningful action when conditions change.
Personnel recovery performance data should be incorporated into EM trending alongside routine monitoring counts. When both data sets are available, the interpretation of environmental trends becomes significantly more robust: a rising count trend accompanied by stable recovery performance points toward an environmental explanation; the same trend accompanied by declining recovery performance points toward a method explanation. Without recovery data, this distinction cannot be made — and the investigation that follows will be inconclusive regardless of how thoroughly it is conducted.
For facilities monitoring water systems and utilities alongside cleanroom surfaces, endotoxin trending data provides an additional investigational dimension. Correlating utility endotoxin results with cleanroom bioburden data enables root cause analyses that consider water system performance as a contributing factor to environmental excursions, an integration that Annex 1’s holistic CCS framework anticipates and that siloed monitoring programs cannot support.6
SOPs, Training, And Culture: Building Competency That Holds
Effective compliance programs are not transplanted from a template; they are built to match the specific challenges, quality maturity, and operational capabilities of the organization they serve. SOPs and training frameworks developed without reference to objective performance data tend to describe ideal technique rather than address the specific failure modes that are actually occurring at a given site.
A well-constructed SOP for surface sampling should do more than describe the procedure. It should define the performance criteria against which the procedure will be assessed, specify the qualification cadence and the triggers for requalification, and establish what happens when an analyst fails to meet the acceptance criterion. These elements transform a procedural document into a quality system component, one that connects daily practice to the CCS and provides the governance structure that Annex 1 provides for.
Training programs that incorporate hands-on assessment with standardized reference surfaces build genuine competency rather than procedural awareness. Analysts who have been assessed against a defined recovery criterion and who understand what their individual recovery percentage means for the reliability of the data they generate, bring a fundamentally different level of ownership to their monitoring responsibilities than those who have simply observed a demonstration and signed a training record.
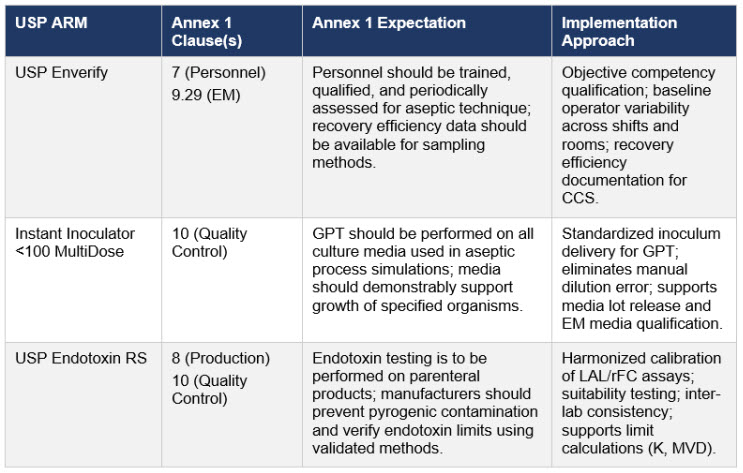
Compliance Mapping: USP Standards Across Annex 1 Expectations
The implementation approaches described below reflect programs developed in direct response to the technical expectations set out in EU GMP Annex 1 and related sterile manufacturing guidance, and are architected around USP Analytical Reference Materials (ARMs) to help ensure that inspection‑facing documentation is grounded in objective measurement data rather than procedural assertion.
Conclusion: From Compliance To Cumulative Assurance
Annex 1’s 2022 revision makes a fundamental point that the pharmaceutical industry can no longer ignore: the subjective, observation-based approaches that characterized EM programs for decades are no longer adequate to support the data-driven contamination control strategies that regulators, patients, and the industry itself require. The potential costs, i.e., missed contamination, misleading trends, inconclusive investigations, and inspection observations, are simply too high.7
The path forward is not about working harder within the existing paradigm. It is about changing the paradigm, replacing observational qualification with quantitative performance data, replacing manual inoculum preparation with standardized reference materials, replacing siloed endotoxin and bioburden programs with integrated monitoring frameworks, and replacing procedural training with competency-based programs grounded in objective measurement.
USP’s ARMs, as well as USP Endotoxin Reference Standard, provide an objective measurement foundation that helps makes this transformation achievable. When recovery efficiency is quantified, inoculum delivery is standardized, and endotoxin testing is anchored to a compendial reference, the data generated by an EM program can be interpreted with confidence, and the contamination control strategy it supports can be demonstrated, not merely described.
The implementation approaches described in this paper were developed through direct collaboration between USP’s microbiology team and Pharmatech Associates’ regulatory and scientific specialists, specifically to operationalize Annex 1 and related guidance using USP’s ARMs as the measurement backbone. The ARMs are not merely supplementary tools that can be bolted onto an existing quality model; in these approaches they serve as the primary measurement foundation around which the implementation framework is built, while still being compatible with and able to enhance existing quality control systems. Gap assessments, EM program design, personnel qualification frameworks, investigation and trending support, and SOP development are all structured around objective performance data rather than procedural assertion and calibrated to each organization’s quality maturity and operational context.
When recovery efficiency is quantified, inoculum delivery is standardized, and endotoxin testing is anchored to a compendial reference, manufacturers can move from subjective EM narratives to defensible, data-driven programs and are better able to demonstrate durable control over the real-world contamination risks that Annex 1 is designed to address.
References
- Parenteral Drug Association. Technical Report No. 13 (Revised): Fundamentals of an Environmental Monitoring Program. 2022.
- BioPhorum. Environmental Monitoring (EM): A Harmonized Risk-Based Approach to Selecting Monitoring Points and Defining Monitoring Plans.
- United States Pharmacopeia. General Chapter <1116> Microbiological Control and Monitoring of Aseptic Processing Environments. USP-NF, 2024.
- Erickson, J. Ensuring Repeatable, Viable Surface Sampling in Aseptic Settings. BioProcess Online, February 14, 2025.
- U.S. FDA. Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice. September 2004.
- United States Pharmacopeia. General Chapter <85> Bacterial Endotoxins Test. USP-NF, 2024.
- European Commission. EU GMP Annex 1: Manufacture of Sterile Medicinal Products. Brussels, 22 August 2022. C(2022) 5938.
About The Author:
 Bikash Chatterjee, chief executive officer at Pharmatech Associates, has 30 years' experience in the design and development of pharmaceutical, biotech, medical device, and in vitro diagnostic products. His work has guided the approval and commercialization of over a dozen new products in the U.S. and Europe. Chatterjee is a member of the USP National Advisory Board and a past chairman of the Golden Gate Chapter of the American Society of Quality. He is the author of Applying Lean Six Sigma in the Pharmaceutical Industry (ISBN-13: 978-0566092046) and a keynote speaker at international conferences. Chatterjee holds a BA in biochemistry and a BS in chemical engineering from the University of California, San Diego.
Bikash Chatterjee, chief executive officer at Pharmatech Associates, has 30 years' experience in the design and development of pharmaceutical, biotech, medical device, and in vitro diagnostic products. His work has guided the approval and commercialization of over a dozen new products in the U.S. and Europe. Chatterjee is a member of the USP National Advisory Board and a past chairman of the Golden Gate Chapter of the American Society of Quality. He is the author of Applying Lean Six Sigma in the Pharmaceutical Industry (ISBN-13: 978-0566092046) and a keynote speaker at international conferences. Chatterjee holds a BA in biochemistry and a BS in chemical engineering from the University of California, San Diego.