
A Checklist For Risk Assessment Of Nitrosamine Impurities In Oral Solid Dose Drugs
By Amol Galande, Ajay Pazhayattil, and Sanjay Sharma

Nitrosamine impurities such as N-nitrosodimethylamine (NDMA) are being identified in high-volume drug products such as valsartan, losartan, irbesartan, and ranitidine at levels well above the acceptable daily intake limits. The credit for identifying the higher NDMA levels goes to an independent pharmacy1 that tests every batch it dispenses, exposing limitations in drug substance and drug product post-approval change assessment and analysis requirements at pharmaceutical manufacturing organizations. Certain continuous process improvement changes (for example, Patent # WO/2011/124655, improved process for preparing valsartan) were deemed as the root cause for the higher amounts of NDMA observed. The issue has resulted in the largest product recalls in recent history, affecting brand and generic products alike. The outcome is a reminder to the industry that lean manufacturing efficiency changes can adversely affect the patient if they do not rely on a sound pharmaceutical science-based technical team performing a 360-degree risk assessment. A rapid reconciliation was required from the manufacturing industry since such incidents raise questions about ICH Q122 adoption readiness. ICH Q12 heavily relies on the robustness and technical strength of a manufacturing organization’s post-approval change management and associated internal quality risk assessment.
FDA Guidance Aims To Control Nitrosamine Impurities
Based on ICH M7 (R1), Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk, N-Nitrosamines are compounds of concern due to their mutagenic carcinogenic effect. Hence, they are to be controlled at or below the acceptable cancer risk levels. Regulators, including the FDA, expect that manufacturers of APIs and drug products take reasonable steps to prevent higher levels or eliminate N-nitrosamines.3, 4, 5 Until recently, nitrosamines were not expected to be formed during the manufacture of most APIs outside the class of sartans with a tetrazole ring. However, based on the new knowledge, the impurities can form during production under certain conditions and when certain solvents, reagents, and other raw materials are used.
Figure 1: Potential Route of Synthesis
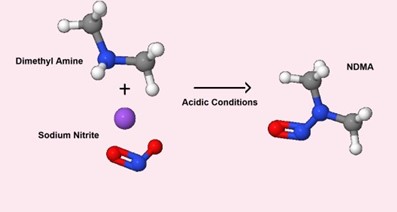
The impurities can also be carried over during the manufacturing process when using specific equipment or some reagents. In cases where nitrosamines can be formed or carried over during manufacturing, the impurities should be controlled or eliminated. The FDA Industry Guidance Control of Nitrosamine Impurities in Human Drugs (Rev 1, Feb. 2021) recommends that drug product manufacturers conduct risk assessments, in collaboration with the drug substance manufacturer, to determine the potential for nitrosamine impurities. The risk assessment should also include evaluation of any pathway (including degradation) that may introduce nitrosamines during drug product manufacture or during storage. If the risk assessment determines that there is no potential for nitrosamine impurities, there is no need to move ahead with further actions. If a risk of nitrosamines in a drug product is identified, then confirmatory testing of batches should be conducted using sensitive and appropriately validated test methods. The regulator has further recommended the risk assessments, the first of three steps (see figure below), be completed by March 31, 2021.
Figure 2: The Three Steps
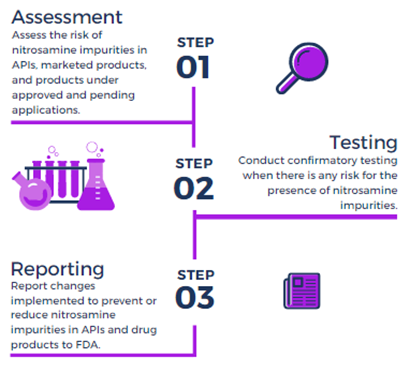
Checklist For Assessing Risk Factors
Table 1 provides a checklist with some factors for a structured initial risk assessment of small molecule oral drug products. Oral delivery routes constitute the largest percentage (~62.02%) of FDA-approved drug product formulations.6 The checklist needs to be adopted only in the context of the product and the manufacturing process being evaluated. The risk factors listed direct the need for careful consideration of the drug substance, excipients, and packaging components, solvents, processing aids, their route of synthesis, manufacturing processes, interactions, degradation, and the derivatives thereof. An equipment evaluation needs to identify the controls in place to preclude the risk of introducing nitrosamine impurities through the use of multipurpose or dedicated equipment. Hence, understanding equipment usage genealogy is important in identifying the risks. The listed factors provided are to be augmented with the available GMP quality system controls, engineering controls (e.g., differential pressure, air changes, AHU filters), and manufacturing controls in drug product manufacturing.
Figure 3: Root Sources for Drug Products
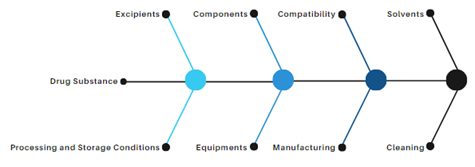
Table 1
|
Risk Factors |
Assessment |
|
Are nitrites (NO2-), nitrous acid, nitrates (NO3-), nitric acid, or azides (N3-) or their sources present in any excipients (e.g., microcrystalline cellulose), processing aids (e.g., water, nitrogen)? |
Base the assessment on the manufacturing process / process flow from vendors, and the declaration report from vendor for proprietary synthetic or non-synthetic routes. |
|
Are peroxides present in any of the excipients, processing aids? |
|
|
Are nitrites (NO2-), nitrous acid, nitrates (NO3-), nitric acid, or azides (N3-) or their sources present in packaging components (including ink, and materials permeability factors)? |
|
|
Is there a risk that secondary or tertiary amine contaminants are present in any primary amines used in your manufacturing process? |
|
|
Are any components containing/potentially containing nitrites and amines present together in solution or in suspension during processing (e.g., during granulation, coating)? |
|
|
Are nitrites (NO2-), nitrous acid, nitrates (NO3-), nitric acid, or azides (N3-) or their sources present in chemically synthesized APIs? |
The drug substance technical justification report for nitrosamine impurities should identify if any nitrosating agent is used/ formed/ carried forward from any source in the manufacturing process. |
|
Based on the structure of drug substance and excipients, is there any possibility of formation of nitroso compounds by interaction of drug substance and excipients? |
Document the structural review to support the conclusion. |
|
Based on the structure of excipients, is there any possibility of formation of nitroso compounds by interaction between excipients? |
|
|
Are any components containing/potentially containing nitrites and amines maintained together at elevated temperatures (e.g., during drying, coating stages, autoclaving, etc.)? |
Review drug product manufacturing processes that involve elevated temperatures. |
|
Do solvents or any other process materials undergo recycling/recovery? |
Review manufacturing processes for use of recovered materials. If used, follow the aforementioned assessment for the recovery steps. |
|
In the manufacturing process of the drug product, are any of the solvents, spent solvents, or process materials treated prior to or during recovery (in-house or by a third party) such that the treatment could lead to formation of amines or nitrosonium ions that could be introduced back into the process through the recovered solvents? |
|
|
Are the recovered materials, if any, dedicated to the process? |
Impacts on other products are to be determined. |
|
Is there a potential for nitrosamine impurity formation during the finished product manufacturing, through degradation and by-products (i.e., if certain excipients, APIs, or packaging components containing sources of amines and nitrite are used together)? |
Review the conditions for degradation/ by-products. |
|
Are there amines and nitrosonium ions (degradation and by-products) likely to come into contact with each other either in the same processing step or through carryover into subsequent processing steps? |
|
|
Is there any potential of nitrosamine formation during storage throughout the finished product’s shelf life? |
Perform literature and synthetic route review to identify if secondary amine formation is possible by the conditions during storage/stability. |
|
Is chloramine used as part of your water treatment, used for cleaning, or as part of the production process? |
Review water treatment and cleaning procedures and materials used. |
|
Have the cleaning solvents/cleaning agents used been assessed for nitrosamine or nitrosamine precursor risk? |
Review cleaning procedures and materials used. |
|
Manufacturing of oral drug product typically involves (e.g., solid oral dry, wet, or direct compression) manufacturing processes utilizing specific equipment. Do any of the processes contribute toward formation of N-Nitrosamines? |
Review manufacturing processing parameters (e.g., RPM) and conditions. |
|
Are sartan drug products manufactured in the same facility? |
Review product portfolio of the formulation facility. |
|
Manufacturing equipment design. |
Review if the equipment meets the current GMP and validation/qualification standards. Confirm continued suitability to the manufacturing and cleaning process. |
|
Manufacturing equipment material of construction. |
The adequacy of the contact surfaces (e.g., SS316, SS316L) and their suitability with respect to the qualified cleaning method, cleaning agent used, and frequency need to be discussed. |
|
Are chemicals such as sodium azide or sodium nitrite, which are primary sources of nitrosamine impurity, used in the facility? |
Review approved materials. |
|
Are cleaning procedures of equipment involved in manufacturing validated using worst-case product consideration (i.e., solubility, potency, toxicity and cleanability)? |
Review cleaning validation master plan, SOPs and reports. |
The initial review for each drug product is expected to provide manufacturers with a high level of confidence on a product or to provide change recommendations for additional controls. As described in ICH Q9,7 the systematic assessment process facilitates science-based decision-making corresponding to the risk observed. The FDA guidance calls for conducting the nitrosamine risk assessment in a timely manner, prioritized based on factors specified in the guidance. Once completed, organizations can move to the second step of conducting confirmatory testing using validated test methods. It is important at this juncture to delineate the responsibilities of such testing in your quality agreement. The newly developed nitrosamine risk assessment report will now be part of documents impacted by upcoming changes, requiring a department to own it.
Conclusion
Despite product recalls, pharmaceutical organizations may not be penalized for the years of patient poisonings on the grounds that they were unintentional. However, any future changes made irresponsibly can be attributed as intentional and may irrefutably impact the organization. The FDA’s nitrosamine guidelines highlight the interventional role of regulatory agencies in protecting the public health by assuring safe drug products. The industry, with its overwhelming acceptance and assessment rates, has yet again shown its capability to quickly align with emerging scenarios while continuing to deliver much-needed drug products to patient populations. That the industry, of which the great majority of volume (~90%) constitutes generic drug products,8 adopted the recommendations within a short window (two years) from the time of finding the carcinogen in a drug product is indeed a tremendous achievement.
References
- https://www.valisure.com/blog/valisure-news/detection-of-ndma-in-raniditine/
- https://database.ich.org/sites/default/files/Q12_Guideline_Step4_2019_1119.pdf
- https://www.fda.gov/drugs/drug-safety-and-availability/information-about-nitrosamine-impurities-medications
- https://www.ema.europa.eu/en/human-regulatory/post-authorisation/referral-procedures/nitrosamine-impurities
- https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/information-health-product/drugs/nitrosamine-impurities.html
- https://www.mdpi.com/1999-4923/10/4/263/htm
- https://database.ich.org/sites/default/files/Q9%20Guideline.pdf
- https://accessiblemeds.org/sites/default/files/2020-09/AAM-2020-Generic-Drug-Biosimilars-Savings-US-Fact-Sheet.pdf
About The Authors:
 Amol Galande, senior manager of the Process Development Lab at Lupin, is an experienced formulation development and process development professional, proficient in regulatory ANDA submissions, especially for generic drug submissions. His oral drug product experience includes R&D, quality, and technology transfer segments with manufacturing organizations in India and Ireland. He has contributed toward bringing affordable generic small molecule drug products to the U.S. and other regulated markets. Galande currently leads a team of process development professionals at one of the world’s largest antibiotic powder for oral suspension manufacturing sites.
Amol Galande, senior manager of the Process Development Lab at Lupin, is an experienced formulation development and process development professional, proficient in regulatory ANDA submissions, especially for generic drug submissions. His oral drug product experience includes R&D, quality, and technology transfer segments with manufacturing organizations in India and Ireland. He has contributed toward bringing affordable generic small molecule drug products to the U.S. and other regulated markets. Galande currently leads a team of process development professionals at one of the world’s largest antibiotic powder for oral suspension manufacturing sites.
 Ajay Pazhayattil is a pharmaceutical management consultant leading technical operations, quality assurance, and regulatory compliance risk mitigation/remediation projects. He has successfully conceived and implemented novel methodologies applying sound pharmaceutical science principles. He is an industrial pharmacist with experience in solid-dose, liquid, and parenteral dosage forms.
Ajay Pazhayattil is a pharmaceutical management consultant leading technical operations, quality assurance, and regulatory compliance risk mitigation/remediation projects. He has successfully conceived and implemented novel methodologies applying sound pharmaceutical science principles. He is an industrial pharmacist with experience in solid-dose, liquid, and parenteral dosage forms.
 Sanjay Sharma, vice president & head of Technology Transfer (Formulations) at Lupin, is a proven pharmaceutical industry leader with years of experience developing, launching and maintaining drug supply. His results-oriented approach and application of technically sound pharmaceutical manufacturing science have enabled some of the largest generic organizations deliver on critical revenue targets. He has identified opportunities applied data-driven decisions in validation and continuous process verification.
Sanjay Sharma, vice president & head of Technology Transfer (Formulations) at Lupin, is a proven pharmaceutical industry leader with years of experience developing, launching and maintaining drug supply. His results-oriented approach and application of technically sound pharmaceutical manufacturing science have enabled some of the largest generic organizations deliver on critical revenue targets. He has identified opportunities applied data-driven decisions in validation and continuous process verification.
Note: This article was prepared by the authors in their personal capacity. The opinions expressed are the authors’ own and do not reflect the view of their employer, government, or any agency with which they are affiliated.