
Assessing pDNA Purity For Cell & Gene Therapies
By Brian Glass, senior analytical consultant, Pharmatech Associates—a USP company

Plasmid DNA (pDNA) is a biomolecule used in vaccines and gene therapies for many types of diseases. One of the basic requirements for materials used in the production of drug products is to assure that the materials are well characterized with respect to purity and impurity profiles of the materials. Small molecules can be characterized using classic analytical techniques; however, determining the impurity profile of biomolecules is more complicated due to their size and structural complexity and because they cannot be constructed via organic synthesis. Because they are produced through cellular processes, the impurity profile of biomolecules is much more intricate, and specialized biochemical techniques are necessary to determine their purity. A recent draft of a proposed chapter1 sheds light on the thinking of the United States Pharmacopeia (USP) regarding this issue. Let’s explore some of the basic types of impurities that should be addressed in a submission and describe some of the analytical technology required for pDNA production when used in a GMP process.
Small molecule therapeutics still prevail as the dominant source of new pharmaceutical products; however, biopharmaceuticals are rising in importance in the pharmaceutical industry and could one day replace small molecules. While the same quality risk management tools used for small molecule drugs can be applied, their application to large biomolecules such as pDNA is more complicated. This article delves into potential testing methods for pDNA to evaluate its suitability for therapeutic applications. It does not aim to be exhaustive but provides a framework for applying development principles of small molecules to large biomolecules.
Limitations Of Small Molecule Analytics
Without question, the workhorse of small molecule analysis lies within the realm of analytical chromatography. High-performance liquid chromatography (HPLC) has emerged as the primary analytical tool for analyzing these compounds. In addition, procedures such as nuclear magnetic resonance (NMR), infrared (FTIR), and mass spectroscopic techniques quickly provide an in-depth determination of the identity and purity of these compounds. In the physical sciences, there is a natural order of progression where we start with more straightforward cases and work our way up to more complex ones. This idea is similar to building a structure: beginning with a doghouse is much more manageable than designing a skyscraper right off the bat.
When it comes to analyzing larger biomolecules, relying solely on small molecule techniques can be challenging and time-consuming. Additional analytical tools are needed to determine the identity, purity, and strength of biologically derived products.
pDNA Identity And Purity Profile
The use of pDNA in the production of biotherapeutics goes back many years. Plasmids are mobile DNA elements that may confer significant evolutionary advantages to the bacterial recipient (e.g., antibiotic resistance). The techniques used to isolate and analyze these compounds in the research lab may need to be revised to ensure the identity and purity of pDNA for human trials. The question is, “How do we produce pDNA for human therapeutic use and ensure that the material is suitable for its intended purpose?” Our focus concerns the word “purity.”
To determine the purity of a small molecule, including the levels and identity of any impurities present, whether known or unknown, it is best to start by analyzing the bill of materials used to synthesize the molecule, the synthetic pathway employed (including reagents, catalysts, etc.), and the purification steps taken to produce the final compound. Figure 1 provides a basic outline of a small molecule API.
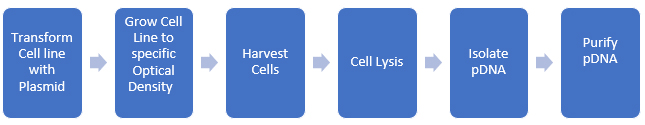
Figure 1: Development of small molecule API
To identify the potential impurity profile, it is crucial to understand the chemistry and processes involved in the early stages of drug development. Through risk analysis and experimental data, organic impurities can be categorized into degradation products, synthetic side reactions, and process impurities. While residual solvents and inorganic impurities are relatively straightforward, organic impurities require additional studies such as forced degradation, structural elucidation, and toxicological assessment of each impurity at or above a determined qualification threshold.
Process impurities and side reaction products can often be reduced through upstream processing techniques such as chromatography, crystallization, and other purification methods. Spike/purge/fate studies can quantitatively demonstrate that these impurities are removed or reduced to acceptable levels. The ICH and USP classify impurities into distinct types, including elemental and inorganic contaminants, residual solvents, and organic impurities, with the latter being the most challenging to determine and manage. We can apply this paradigm to biomolecules as well. pDNA is produced in bacterial cell lines and then isolated and purified. A rough schematic of the process is shown in Figure 2.
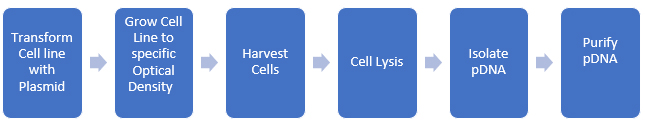
Figure 2: Development of pDNA for use in biopharmaceuticals
At each step, we need to evaluate potential impurities, and there are many more steps required to produce purified pDNA than there are for small molecules (usually). Additionally, we should consider metabolic impurities the cell line produces during growth. Since we are using the cell’s biological systems to make the biomolecule, the level of complexity of the synthesis increases enormously (as does the potential impurity profile)!
Assessing Risk Associated With Impurities
During the process of developing analytical procedures to detect and quantify impurities in pDNA, it is important to assess the level of risk associated with each impurity. The acceptable levels of impurities may vary depending on the intended use of the pDNA. Quantifying the level of impurities that would lead to certain levels of risk can help to establish appropriate limits for impurities. Such quantification can be achieved through risk analysis and experimental data.
Low levels of host cell DNA, RNA, and protein do end up in the final product, but it’s important to determine, based on the application, what is acceptable and the risk level. Significant attention is needed to ensure that the pDNA produced meets the critical quality attributes (CQAs) previously established. It can be reasonably expected that the impurity profile will include small (antibiotics, etc.) and large molecules (host cell DNA, etc.).
The last step will be the final testing and release of the pDNA and generation of the Certificate of Analysis (COA), which will be required by manufacturing facilities utilizing the product. It is essential to ensure that the COA for pDNA is populated with data collected with full validation analytical procedures. Table 1 outlines some of the possible tests that require consideration for pDNA. However, additional tests may be necessary depending on the specific use of the pDNA and its exact specifications. It is essential to perform all the tests required to ensure that the pDNA is suitable for its intended purpose and meets all regulatory requirements before commercialization.
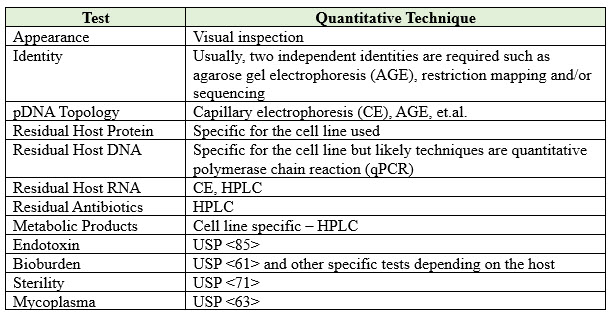
Table 1: Possible COA for pDNA
It is important to pay attention to the endotoxin levels if the plasmid is replicated in a gram-negative host cell like E. coli. Additional processing and purification steps may be required to reduce endotoxin to acceptable levels. It is also important to evaluate the presence of mycoplasma as a potential impurity.
Summing Up
The draft USP guidance <1040>,1 which is available for comment in the Pharmacopeial Forum, mentions these and other tests required for quality considerations of plasmid DNA as a starting material for cell and gene therapies. Although it may never become official, it provides USP's current thinking based on collaboration with cell and gene therapy industry experts. To ensure the production of a high-quality product, one must develop a solid understanding of all components used in the manufacturing and purification process of the final product. Each item used is a potential impurity, and materials used in the production of pDNA (or any similar biotherapeutic) should be sourced from validated suppliers, and the impurities present should be understood and assessed. While it is unlikely that suppliers of biotechnology products that produce pDNA have a Drug Master File (Type I or Type IV, depending on the use), it would be advantageous if a supplier provided this level of detail. In summary, producing a biomolecule such as pDNA is still a process, and some of the techniques used for small molecule APIs can still be applied.
Reference
- 〈1040〉 Quality Considerations of Plasmid DNA as a Starting Material for Cell and Gene Therapies. The Biologics Monographs 5—Advanced Therapies Expert Committee is proposing the following new general chapter. The goal of this general chapter is to describe considerations for the manufacture and release of plasmids for use as starting materials in the manufacture of cell and gene therapy drug products. Because plasmid DNA suppliers have been taking different approaches to manufacturing and applying different specifications, this general chapter was developed to assist manufacturers of gene therapy drug substances and drug products in building a control strategy for the plasmid DNA used as a starting material.; (BIO5: R. Potts); Correspondence Number — C320084; https://doi.usp.org/USPNF/USPNF_M17795_10101_01.html.
About The Author:
 Brian Glass, senior analytical consultant at Pharmatech Associates, has over 30 years’ experience in the fields of analytical research and development, quality control, validation, technical transfer services, and process development. He has held senior analytical positions in private startups, established pharmaceutical manufacturing facilities, and large CMO/CDMO organizations. His career spans all facets of the drug development process for small- and large-molecule therapeutics, from preclinical to commercial products, utilizing different modes of drug delivery systems (solid dosage, injectable, inhalants, oral solutions/suspensions, and soft gel capsules). He holds a B.S. in zoology from Louisiana Tech University.
Brian Glass, senior analytical consultant at Pharmatech Associates, has over 30 years’ experience in the fields of analytical research and development, quality control, validation, technical transfer services, and process development. He has held senior analytical positions in private startups, established pharmaceutical manufacturing facilities, and large CMO/CDMO organizations. His career spans all facets of the drug development process for small- and large-molecule therapeutics, from preclinical to commercial products, utilizing different modes of drug delivery systems (solid dosage, injectable, inhalants, oral solutions/suspensions, and soft gel capsules). He holds a B.S. in zoology from Louisiana Tech University.