
Continued Process Verification And The Validation Of Informatics Systems For Pharmaceutical Processing
By Gloria Gadea-Lopez (Shire), Rob Dimitri (Shire), Rob Eames (GSK), Peter Hackel (Shire), Eric Hamann (Pfizer), Robin Payne (BioPhorum), Carly Cox (Pfizer), and Steve Kane (Shire)
Continued process verification (CPV) is an activity that provides ongoing verification of the performance of a manufacturing process. Guidance issued by the FDA in 20111 emphasized the importance that manufacturers engage in CPV as an integral part of their process validation life cycle. It provides the manufacturer with assurance that a process remains in a validated state during the routine manufacturing phase of the product life cycle.
The BioPhorum Operations Group published an industry position paper on CPV for biologics2 in 2014. BioPhorum members then published a road map (an accessible step-by-step guide) for the implementation of CPV3 in 2016. The team also published a paper on CPV for legacy biopharmaceutical products in 2017.4
CPV is fundamentally a formal means by which the performance of a commercial manufacturing process is monitored to ensure consistently acceptable product quality. It includes preparation of a written plan for monitoring a manufacturing process, regular analysis of results, documentation of the data collected, and analysis of data and actions taken based on the results of monitoring the process. Some elements of the CPV data set are likely to overlap with existing GMP systems, such as those related to batch release (BR) decisions, annual product review (APR), and change control. Despite the overlap with data used for BR, the CPV system is separate from and typically does not influence batch release decisions. This separation is extremely important, because it reduces the level of business risk associated with the CPV informatics (CPV-I) system and hence the criticality of the validation procedures.
The volume of data required to deliver CPV is large and involves a complex landscape of data sources and data integrations (see Figure 1). This makes the deployment of informatics systems, and automated data gathering and analysis, important if speedy and efficient process control is to take place. This article, written by members of the BPOG CPV Informatics team, summarizes their collective experience in validating the informatics components of their CPV programs, to shed light on common issues and provide recommendations and best practices. It covers the typical scope of CPV-I validation, validation strategies, the importance of system design, data integrity and change management. The topics are presented in the context of the risk-based approach to validation advised by the FDA and embodied in the good automation manufacturing practice (GAMP) guidance published by the International Society of Pharmaceutical Engineers (ISPE).5 The full paper that this article is based on is available on the BioPhorum website.6
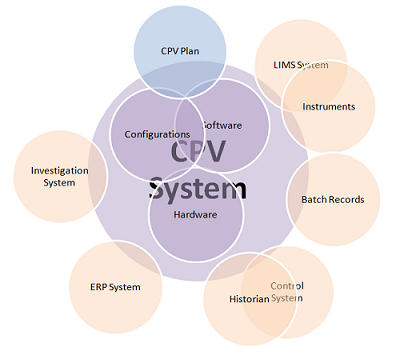
Figure 1
Validation Scope And Strategy
Common elements to be included in the validation of a CPV-I solution are highlighted in Figure 2. In reviewing the scope of the validation, a reasonable approach is to refer to such a graphic as a way to gain consensus among various stakeholders in the CPV program. Starting here allows the stakeholders to customize the high-level diagram with the specific details for each product.
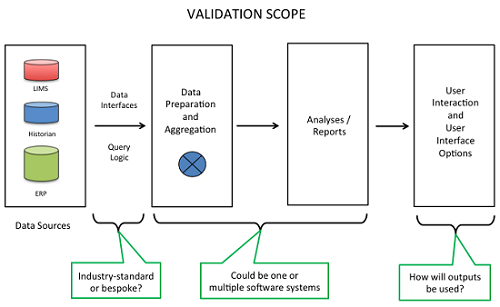
Figure 2
When reviewing the data sources, key questions to ask include: are the source data systems validated; is required data accessible in electronic format; will manual data entry be required; does the data to support the program span multiple data sources and if so, how will the data be aggregated together?
The interfaces between systems are critical focus areas for validation because of the risks for data integrity. Whenever multiple systems “talk” to each other, there is a risk for data transfer errors such as an improper data format being applied to a stream of data. A key initial question to begin to highlight risks is whether the required interfaces are off-the-shelf and already in use, or do they need to be bespoke designed and implemented. The bespoke approach is more risky with regard to data integrity, so appropriate levels of due diligence and verification are required.
Another key discussion topic is whether the data preparation and aggregation functions will be performed by the same or separate software systems as the analyses and report generating functions. There are pros and cons with either approach. If the same system performs these functions, it could simplify the validation, whereas the advantage of using multiple systems is that it allows for a best-of-breed approach.
Typically, a system generates graphical and tabular outputs. These may be produced manually or, more typically, run via automation. From a validation perspective, configurations that run via automation are less risky since they can be designed and verified and then used repeatedly. If the system is commercial-off-the-shelf software, then the vendor’s guarantee that its operations are reliable can be leveraged.
It is also important to evaluate all the possible options that users will need in working with the outputs from the CPV-I solution. For each user scenario, such as accessing the data or performing follow-on analyses, the functions may become the definition of testable design elements with their respective validation test cases to demonstrate that the capabilities meet their intended uses.
There are many different ways to approach the validation strategy, depending on the scope of intended uses and the system design. As shown in Figure 3, many pharmaceutical manufacturers view CPV as a global general service, resulting in a harmonized suite of capabilities across sites and a global shared services model with standard offerings, standard interfaces, a library of configurable components, and a support model.
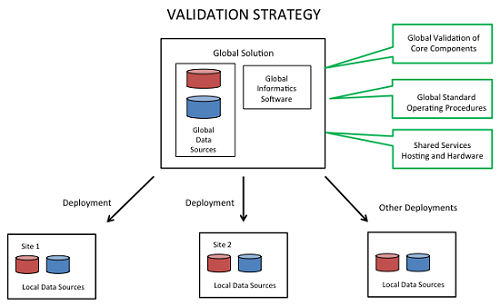
Figure 3
A global strategy has a number of advantages, including streamlined validation. It allows, for example, global validation of core components and the use of reusable documentation, including referenced validation packages and SOPs.
Data Integrity
Regulatory agencies emphasize the importance of data integrity and have published recent guidance stressing its importance to a CPV program. The main challenge, as illustrated in Figure 4, is to ensure that data used for CPV is complete (i.e., no missing data), accurate (i.e., they are the approved results and are assigned to the correct attribute or parameters), precise (i.e., managed and presented to the number of decimal places appropriate for the respective measurement system), and presented in context (i.e., associated to the correct batch number, date, time, etc.).
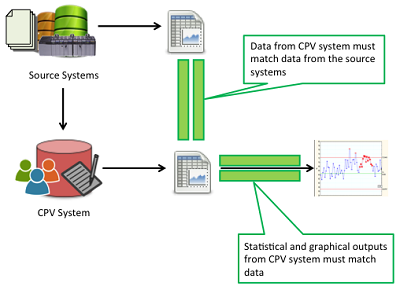
Figure 4
Challenges to data integrity include: data is sourced from a variety of systems that are managed by multiple owners; data is used in a variety of ways to support CPV; data entry may be manual; and the number of data elements monitored in a CPV program can be large. Given the complexity and volume of data streams in a CPV program and the multitude of opportunities for data integrity issues, how is data integrity ensured?
First, data integrity must be a part of a company’s culture. That cultural component is necessary because so many people will be involved in the entry, management, and support of the results. Creating this data integrity culture is partially the mandate of the leadership/management team at an organization and also the result of effective training (both onboarding and periodic refresher training) and work aids.
Other key components to data integrity include: change management, as data sources and CPV programs change over time; proper definition of user requirements and design of the CPV-I solution; cross-functional review of all the data sources, data interfaces and data aggregation/preprocessing as part of the design; and the level of automation, as manual human intervention increases the risk for error in data integrity. More detail on ensuring data integrity is provided in the full paper.6
System Design
The world is constantly changing, and pharmaceutical manufacturing is no different. Once a validated Informatics solution is delivered to support a CPV program for one or more products, things change, and the system must be designed to support that change while maintaining its validated state.
Typical changes that will cause an impact include: refinement of the control strategy and CPV protocol as more experience is gained with the process; changes to batch documents/recipes and equipment; changes to data sources, data types, and scenarios; changes to analytic methods and in-process measurements; and changes to supporting IT systems and platforms.
Given the likelihood of future changes (both expected and unexpected), a best practice is to design the system configurations for flexibility without required rework and revalidation. Note that the specifics for how to do so depend on the specific systems and informatics components in use to deliver the CPV program. Different systems will have different approaches and capabilities for designing them for configurability. The general design principles to consider include:
- The use of a common, shared core set of configurations to deliver the baseline CPV functionality across product attributes and across multiple products. Optimally, the core shared configuration would be capable of delivering the relevant scope of functions required by all products’ parameters and attributes, with the capability to dial in the specific configuration properties to fit the specific needs for each product and attribute. With this approach, there is less complexity ongoing over time to document, test, and manage.
- The use of lookup tables, where each core informatics configuration looks up its instructions at run-time. One advantage of this approach is that ongoing changes are easier to implement, since they are not buried in the details of the configurations themselves and since the use of the lookup table approach was verified and validated as an intended design of the solution.
- The use of integrated versioning and history, where if a change is made to a configuration, the old version is retained (e.g., as version “1.0”) and the new one is defined as the current version in use (e.g., as version “1.1”). Integrated versioning and history streamlines the ongoing validation as changes are deployed because the system is self-documenting and provides inherent capabilities for rolling back to previous versions.
Up-front investments in the future-oriented design of a CPV informatics program pay dividends over time. It is important to establish from the start whether a team is designing an approach for one or more specific products or as a general solution across many products. If the latter, and a holistic approach is taken in assessing the requirements and design details, a great deal can be streamlined by designing and configuring once and reusing again and again. In this case, specific general capabilities and intended use cases can be documented and tested in an overall master validation exercise.
System Testing
The focus here is on the effective and optimized testing of a CPV-I solution. Risk-based validation is a common approach to direct the validation to the high-risk and high-impact intended uses and system components. This is consistent with FDA guidance and ICH Q8.7 It is effective because it takes into consideration a systematic approach to leverage the subject matter expertise of the most knowledgeable stakeholders in your organization. In short, the stakeholders of the CPV program specific to a product rank the items that are most important. The most important and most risky functions receive the most scrutiny and testing. The least important and least risky functions receive commensurate scrutiny and testing. Given the size and scale of a CPV program, an exhaustive verification is not feasible. Instead, a risk-based approach is an optimal way to allocate time and resources in a deployment to maximize their effectiveness and to avoid non-value-added work.
What does a risk-based approach look like? Stakeholders are selected and their subject matter expertise is taken as input into the validation plan that provides the details of the validation scope, approach, and acceptance criteria. Typically, an organization will have a standard risk assessment template (see Figure 5 for the typical inputs into a risk assessment matrix) to provide structure and a common approach to gathering and agreeing on this input.
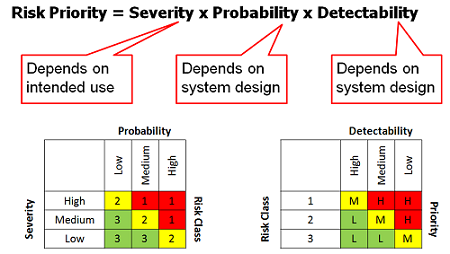
Figure 5
Key success factors for a risk-based approach include: assembling the complete set of stakeholders engaged in the process for gathering and assessing risks; taking an iterative approach to ensure adequate feedback into the risk assessment and the team’s agreement on the risk scores; and getting support from the validation and quality professionals that the risk scores will be used to focus the validation scope and efforts.
Note that using commercial-off-the-shelf software has a number of advantages, including streamlined validation. The vendor often provides a validation package that can be leveraged, as can its quality management system (QMS). Automatic testing can also streamline validation.
Change Management
This section deals with maintaining the validated state of a CPV-I system, given that some elements of the system will be subject to frequent change. When designing the informatics aspects of a CPV program, it is important to plan for change, as outlined above, and plan for managing those changes.
A good starting point for this is to invest in a map of the people, processes, and systems that touch the CPV program to identify the touch points and potential areas impacted by potential changes. Based on those touch points, identify the existing governance processes for evaluating and responding to changes that will impact the informatics components of your CPV program. A subset of the governance processes will be specific to the CPV-I system and configurations itself, and it is important to use the input from the program’s risk assessment to define the levels of categories of configurations and the processes by which they will be managed.
Awareness of changes is a key aspect of managing change, and the map will greatly assist in that. It will pay dividends to brief stakeholders on the scope of attributes that are going to be monitored, the way data is collected and managed, and the flow of information in the system. It will help your colleagues understand and support the CPV efforts by understanding how changes may impact the informatics aspects of the program. It is useful to leverage subject matter experts’ guidance about the change management controls and processes governing their respective areas. For the changes that may impact the CPV-I solution, it is important to explore the potential for notifications, such as a notification step in the respective change control process.
With touch points between systems enumerated and notification systems in place, a next layer includes the definition of business processes with roles and responsibilities for managing changes. When a specific change notification is raised, managing the change includes the definition of ways of working to influence the timing or scope of those changes.
There are many things that can be implemented from the start to make changes easier to manage from a validation perspective. Many or all of these practices are outlined in the relevant GAMP validation standards.
One fundamental planning decision is the informatics computer systems architecture. Without going into detail, it is very helpful to set up separate environments for informatics systems, including development, test, and production.
Another fundamental foundation for change is the initial validation documentation set, which often includes: installation plans, requirements specifications, configuration specifications, test cases, user training, user SOPs, and the system administrator SOP. The core documentation set becomes a foundation to streamline the ongoing management of changes. The system administrator SOP, for instance, provides important guidance on how to perform changes and ensures that they are implemented in a consistent and compliant way. The set should be considered a living documentation package to be augmented as additional requirements or intended uses arise.
Finally, as changes in requirements and data sources occur and software version upgrades happen over time, there will be a need to re-execute testing. Re-executing previously executed test cases to ensure current conformance to expected results is called regression testing. Using a risk-based approach combined with a comprehensive impact assessment of the changes (e.g., a vendor’s release notes in the case of a software upgrade) allows for the sub-setting of an entire suite of test cases to select ones relevant to the specific changes.
Summary
Guidance issued by the FDA has emphasized the importance of manufacturers engaging in CPV as an integral part of their process validation life cycle, as doing so provides the manufacturer with assurance that a process remains in a validated state during the routine manufacturing phase of the product life cycle. The volume of data required to deliver CPV is large and involves a complex landscape of data sources and integrations. This makes the deployment of informatics systems, and automated data gathering and analysis, important to enable speedy and efficient process control.
References:
- Guidance for Industry, Process Validation: General Principles and Practices, US Food and Drug Administration (2011), http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
- Continued Process Verification: An Industry Position Paper with Example Plan, BioPhorum Operations Group (2014). https://www.biophorum.com/download/cvp-case-study-interactive-version/
- Boyer M, Gampfer J, Zamamiri A, et al. A Roadmap for the Implementation of Continued Process Verification, PDA J Pharm Sci and Tech 2016, 70 282-292.
- https://www.biophorum.com/download/continued-process-verification-cpv-of-legacy-products-in-the-biopharmaceutical-industry/– ‘Continued Process Verification of Legacy Products in the Biopharmaceutical Industry’]
- GAMP® 5: A Risk-Based Approach to Compliant GxP Computerized Systems, http://www.ispe.org/gamp-5#sthash.h0zFHOa2.dpuf
- https://www.biophorum.com/download/continued-process-verification-and-the-validation-of-informatics-systems/ – ‘Continued Process Verification and the Validation of Informatics Systems’]
- International Congress on Harmonization (ICH) Q8, https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf