
Commissioner Gottlieb, CAR T-Cells, & The Future Of Rare Disease Therapies
By Jill Hartzler Warner, Catalyst Healthcare Consulting, Inc.
FDA Commissioner Scott Gottlieb, M.D., plans to further incentivize development of therapies for rare diseases and targeted therapies by making it a top priority for this administration. He has taken the wheel at an exciting time, when insights from genomics are starting to come to fruition and promising new therapies based on new technologies are being developed. This article explores some of the policy changes happening now in the regulatory rare disease space — and shifts we expect to see in the future — as the FDA’s leadership team implements these changes at the agency.
Commissioner Gottlieb’s Priorities For Change
In an FDA Voice blog post this June, Dr. Gottlieb wrote:
It is incumbent upon FDA to ensure that we have the right policies in place to promote and encourage safe and effective innovation that can benefit consumers, and adopt regulatory approaches to enable the efficient development of these technologies. By taking an efficient, risk-based approach to our regulation, FDA can promote health through the creation of more new and beneficial medical technologies. We can also help reduce the development costs for these innovations by making sure that our own policies and tools are modern and efficient, giving entrepreneurs more opportunities to develop products that can benefit people’s lives.
This quote highlights several important themes now playing out across multiple areas of FDA responsibility:
- Revisiting and modernizing FDA policies to advance and appropriately review innovative technologies.
- Embracing risk-based policies with an eye towards making them more efficient and reducing unnecessary costs that come with bringing targeted products to market.
- Looking for new ways FDA might appropriately infuse greater competition in the marketplace.
As this new commissioner begins to set the course for the agency, he seems to be carefully harnessing the strengths of new and existing authorities. He is asking his team to take advantage of the new tools and pathways Congress provided in the 21st Century Cures law and in the FDA Reauthorization Act, while crafting organizational changes that will help advance his vision.
To meet the challenges of new therapies that do not fit neatly into therapeutic silos, Dr. Gottlieb is leveraging cross-cutting management structures such as the Oncology Center of Excellence (OCE) to enhance coordination within the agency. At the same time, he is engaging frequently with the public, and we expect his leadership team will increasingly collaborate with academics, researchers, patients, and other stakeholders to tackle broader issues in which the agency is but one of many players.
Innovation Plan Coming Soon
Dr. Gottlieb is touting the soon-to-be-released Medical Innovation Development Plan as his blueprint for the changes he wants his team to pursue to foster innovation across FDA’s medical product centers. It seems like he has been sprinkling his recent series of speeches with components of the plan. Here are a few noteworthy ones from the orphan drug community perspective, to help reduce cost of development and thereby speed the development of promising therapies:
- Recognizing platforms: Dr. Gottlieb believes that both sponsors and the agency can be more efficient when products that share a common “platform” also share the learnings across the technology. Cell and gene therapy, regenerative medicine, and other technologies that target the molecular and genetic basis of disease may be some early areas where considering a platform approach makes sense. Though this is not a new concept, new provisions in the Cures legislation clarify the agency’s authority, in certain cases, to leverage data from previously approved applications for genetically targeted drugs and variant protein-targeted drugs for serious or life-threatening rare diseases.
For those who have not always followed the FDA, the agency hasn’t always embraced the idea of platforms. We’ll be watching how it rolls out this approach. Notably, the agency’s Cures work plan calls for new experts to support the review of targeted therapies. And part of the plan is to make early feedback meetings more accessible, so sponsors can avoid unnecessary preclinical work that can be costly.
- Modernizing how clinical information is collected and how it is evaluated and reviewed: Dr. Gottlieb is taking note of the increasing cost of early-stage drug development, which is rising faster than in other segments. We expect to see the agency using new tools and methods to increase efficiency, and encouraging sponsors to do so as well.
- Increased opportunities to communicate with review staff for new technologies: Many of the changes will require clear communication with sponsors for successful adoption. We expect the early engagement feature of accelerated pathways such as the Regenerative Medicine Advanced Therapy (RMAT) designation (discussed below) to pave the way for innovative therapies.
- Aggressive guidance plan: The FDA needs more rapid policy development, including guidance documents, to ensure that product sponsors receive the most up-to-date advice, according to Dr. Gottlieb and other key agency leaders. The agency will be busy with work on at least 10 new disease-specific guidance documents over the next year. Although these will address both common and rare diseases, stakeholders will want to stay tuned for more announcements. FDA is already drafting guidance on amyotrophic lateral sclerosis and one on Alzheimer’s disease.
- Organizational changes: To support his forward leaning plan, policy and structural changes to increase cross-agency coordination are a likely outcome. Though it has been in the works for over a year, the Center for Drug Evaluation and Research’s (CDER’s) Office of New Drugs reorganization continues to move forward. We anticipate that it may morph from a division-by-division staffing model to a model where staff would move between review divisions as needs arise — and with encouragement for division directors to be more outwardly focused, to better interact with stakeholders, collaborators, and other regulators.
Dr. Gottlieb has indicated that he wants change in how the agency communicates with stakeholders. In a recent speech, he emphasized that this is not “business as usual” and that the new approaches may “require a much more iterative process.” A new pilot program is expected to provide one such opportunity for sponsors to collaborate with the agency.
How Will FDA Implement New Incentive Pathways (And Modify Others)?
Another area that sponsors should watch is the FDA’s implementation of new designation and incentive pathways in the Cures legislation that promise to speed development for eligible products.
Regenerative Advanced Therapy (RMAT) Designation
The RMAT designation adds to the array of designation and priority pathways in the Federal Food, Drug, and Cosmetic Act (FD&C Act), and knowing when and where to apply for these can bring significant advantages to sponsors.
Regenerative medicine therapies for serious or life-threatening diseases for unmet medical needs — and that have promising preliminary clinical evidence — are eligible for designation as a “regenerative advanced therapy.”
Winning The RMAT Prize
Like breakthrough therapy designation, RMAT designation gives sponsors valuable benefits: notably, early access to FDA and personalized guidance on product development to expedite review. The product application may be eligible for priority review and accelerated approval, and certain flexibilities in premarket and postmarket data may apply.
We anticipate that the agency will use its discretion in interpreting the Cures provision to include a broad scope of products. Dr. Gottlieb and other agency leaders have already announced that at least some gene therapy technologies, such as chimeric antigen receptors (CAR) T-cell therapies, will be included. While the FDA’s Center for Biologics Evaluation and Research (CBER) is actively reviewing and deciding designation requests, stakeholders would benefit from greater clarity about what types of products will be included. Sponsors should be thinking now about this important pathway, and we expect to hear more about RMAT soon — possibly within the broader Medical Innovation Development Plan.
Overhaul Of Orphan Designation?
While FDA is expanding incentives for product development on the one hand, we are closely watching signs that the agency may narrow which products qualify for orphan designation — a program that offers user fee waivers, tax credits, and potential 7-year market exclusivity, among other benefits, for eligible therapies. Targeted therapies put pressure on the current orphan designation policies, as “common” diseases can qualify as orphans when factors (for example, mechanism of action) limit their use to a smaller population. Dr. Gottlieb appears to be reviewing policies that some view as “gaming the system,” and we may see changes intended to promote competition and close loopholes.
At the same time, Dr. Gottlieb has demonstrated support for the orphan designation program, and has already implemented efficiencies to eliminate a backlog of designation requests, which have grown dramatically in recent years while staffing has stayed flat. He’s pledged to review all future requests within 90 days, and a new designation template should add clarity to the process.
Pricing
In the past, the FDA has stayed away from the issue of drug pricing since it is not traditionally within the agency’s purview. This has benefited rare disease treatment innovations that have had high price tags due to the typically long development path and narrow band of patients who can use the products. Dr. Gottlieb has deftly finessed the issue. He has answered the calls for the FDA to help drive down the price of drugs by setting priorities that will enhance generic competition, increase the efficiency of product development without sacrificing safety and efficacy standards, and close loopholes that can create unfair monopolies.
Kymriah: A Promising Example
Just a few weeks ago, the FDA approved the first gene therapy in the U.S. Kymriah is a CAR T-cell gene therapy for a form of acute lymphoblastic leukemia (ALL) in children and young adults who have failed other treatments, a rare indication. Stakeholders, industry, and investors have marked this as a bellwether for the likely approvals of other targeted therapies.
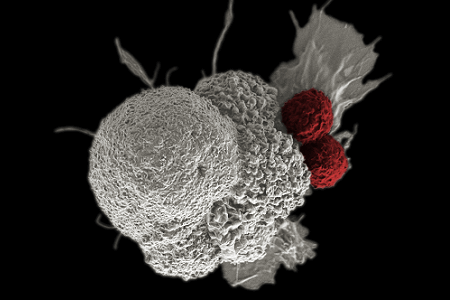
Photo of immune cells activated by CAR T-cell therapy attacking cancer cells (courtesy of National Cancer Institute)
The good news for the orphan drug community:
-
The FDA acted remarkably quickly, approving Kymriah a month before its Prescription Drug User Fee Act (PDUFA) date and only seven weeks after the Oncologic Drugs Advisory Committee voted unanimously to approve it.
-
It was a good example of cross-cutting coordination to bring the right experts to bear on review of a novel technology. While Kymriah is regulated by CBER, the OCE led the clinical review. The OCE, which draws from experts in CDER, the Center for Devices and Radiological Health (CDRH), and CBER, was created to provide an integrated approach to review of cancer therapies and expedite their development. CBER addressed all other aspects of the review and made the final approval decision.
-
The approval also demonstrated the FDA’s willingness to understand the unique nature of a new technology and shift the agency’s traditional premarket focus to a lifecycle approach. As advisory committee members discussed, Kymriah has a remarkably effective profile for patients for whom death is almost certain without treatment; however, it also carries significant safety risks. FDA recognized that the gene therapy technology brings a different set of issues than traditional drugs that are more readily characterized, and approved the product swiftly but with postmarket safety requirements, including a postmarket observational study.
Where The Rubber Meets the Road
The initiatives are ambitious. How far and how deeply Dr. Gottlieb will be able to effect real change at the agency remains up in the air. The FDA is a huge 18-wheeler that is difficult to steer, flying down the highway, and the new commissioner’s success will depend on his ability to create institutional change at many levels.
One way FDA commissioners attempt to translate their efforts from “podium policy” to action is by incorporating the new policies and procedures into guidance and other documents. When policies and procedures are in guidance, staff members are generally required to follow them, and they are in place for future reviewers as well. The process of guidance development also has the benefit of offering opportunities for stakeholder input — well-crafted comments can influence the content of the final document.
One challenge we see for this commissioner in leaning heavily on new guidance is that it is at odds with the Trump administration’s “2-for-1” order, which requires that for every regulation or guidance that the agency promulgates, it must withdraw two. There are some exceptions and ways around the order, but regulatory actions required by law — such as guidance the Cures law earmarks for FDA’s to-do list — are not automatically exempted. And the new requirements for economic analyses in guidance documents will likely delay their development. Although the agency is implementing the RMAT designation now — and the FDA is not required to draft guidance — which products will qualify for the designation is still somewhat uncertain, and many stakeholders are looking to agency guidance to clarify eligibility.
Dr. Gottlieb has signaled his support for increased opportunities for sponsors with innovative products or methods and patient advocates to collaborate with the FDA. We’ve already seen in the approval of Kymriah that the agency can act swiftly to assess the risks and benefits, and establish the appropriate controls to make an innovative, effective treatment for a devastating disease available to patients. This approval sets the stage for other products in the pipeline, and sponsors will do well to pay attention.
Dr. Gottlieb’s statements and actions to date are encouraging steps for the many patients with rare and ultra-rare diseases. He is a doctor and FDA veteran who has himself battled cancer, and his focus on innovation for cures is timely and needed. Companies with rare disease pipelines are advised to stay ahead of the many expected policy changes that can help bring safe and effective products to the patients who need them.
Next Steps
Dr. Gottlieb will speak about innovation in the postmarket phase of product development at the end of September. Gene therapy products are likely to be a particular focus of Dr. Gottlieb’s plans, because they have sustained effects that will need to be monitored long-term. One key challenge is how to follow patients for potentially a lifetime without overly intruding upon their lives. The FDA is never short of challenges to overcome, but the race is on, and the finish line is not yet in sight.
About The Author:
 Jill Hartzler Warner, J.D., is VP of regulatory policy at Catalyst Healthcare Consulting, Inc. She is an expert in strategic regulatory positioning, legal analysis, policy development, and problem solving. From her 30-plus-year career at FDA, she brings her depth and breadth of experience and understanding of agency approaches gleaned from her many leadership roles at the agency, including associate commissioner, senior advisor, and associate chief counsel.
Jill Hartzler Warner, J.D., is VP of regulatory policy at Catalyst Healthcare Consulting, Inc. She is an expert in strategic regulatory positioning, legal analysis, policy development, and problem solving. From her 30-plus-year career at FDA, she brings her depth and breadth of experience and understanding of agency approaches gleaned from her many leadership roles at the agency, including associate commissioner, senior advisor, and associate chief counsel.
During her diverse career at FDA, Ms. Warner was responsible for executive oversight of the agency’s offices of orphan products development, combination products, and pediatric therapeutics, among others. She provided key input on significant FDA legislation, including the 21st Century Cures Act. As lead architect and author of FDA’s approach to cell and tissue product regulation, she brings significant expertise in regulation of regenerative medicine.
Catalyst Healthcare Consulting, Inc. is a strategic policy and regulatory affairs advisory firm. Catalyst partners with biopharma, medical device, combination product, digital health, venture capital, trade groups, and patient advocacy clients to distill complex healthcare issues into meaningful commercial and philanthropic opportunities.