
Combination Products Vs. Combination Therapies: What Is The Difference?
By Lynn Hansen, Pharmatech Associates

While the designation for what constitutes a combination product is clearly defined by the FDA, industry continues to push the boundaries for disease treatment with new and innovative breakthrough drug therapies. As regulatory professionals, the challenge we face is to align these new therapies with application-filing strategies including investigational new drug applications, new drug applications, biologics license applications, and medical devices.
Regulatory Pathway For Combination Products
Combination products are clearly defined in 21 CFR 3.2(e), and the Office of Combination Products has a charter in place. Various offices within the FDA have identified combination treatments, therapies, and regimens in recent approvals.1 Strictly speaking, these approvals are not combination products because they are drug/drug or biologic/biologic combinations that — when combined — aid in the treatment of specific indications. From a regulatory standpoint, there may be instances where combination treatments fit in with combination products. However, unless the therapies meet the criteria identified in 21 CFR 3.2(e), the treatment is not a combination product in the eyes of the agency.
There are four main categories for combination products:
- a product composed of two or more regulated components (i.e., drug/device, biologic/device, drug/biologic, or drug/device/biologic) that are physically, chemically, or otherwise combined or mixed and produced as a single entity
- two or more separate products packaged together in a single package or as a unit and composed of drug and device products, device and biological products, or biological and drug products
- a drug, device, or biological product packaged separately that, according to its investigational plan or proposed labeling, is intended for use only with an approved individually specified drug, device, or biological product. Furthermore, both are required to achieve the intended use, indication, or effect and whereupon approval of the proposed product, the labeling of the approved product would need to be changed (e.g., to reflect a change in intended use, dosage form, strength, route of administration, or significant change in dose)
- any investigational drug, device, or biological product packaged separately that, according to its proposed labeling, is for use only with another individually specified investigational drug, device, or biological product where both are required to achieve the intended use, indication, or effect.
Evolution Of Combined Treatments
As technology introduces new combination products, a number of new combined treatments, therapies, and regimens have been tested side-by-side and proven to help patient populations. Many of these early combination treatments are regulated by the FDA’s Center for Biologics Evaluation and Research (CBER) for use in patients at the end of known beneficial dosing. Some approvals have been granted specifically for patients with certain types of high-risk acute myeloid leukemia (AML), combining two commonly used chemotherapies into a single formulation that may help patients live longer than if they received the two therapies separately. According to Robert Peter Gale of the Imperial College London, the reason for this is because “cancer drugs are most effective when given in combination. Sometimes combination drug therapy is used not to cure but to reduce symptoms and prolong life.”
FDASIA Breakthrough Therapy Designation
It is likely the terms “combination treatments/therapies/regimens” came into use because of the July 9, 2012, enactment of the FDA Safety and Innovation Act (FDASIA) that expanded the agency’s authority for designating breakthrough therapies.2 For our purposes, the definition of a breakthrough therapy is a drug:
- intended alone or in combination with one or more other drugs to treat serious or life-threatening disease or condition
- for which preliminary clinical evidence indicates the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial effects observed early in clinical trials development.
The FDA will expedite the development and review of a drug designated as breakthrough therapy. This means all requests for breakthrough therapy designation will be reviewed within 60 days of receipt, and the FDA will either grant or deny the request. Since the FDASIA was instituted, an increase in requesting breakthrough therapy designation has erupted, especially in the FDA’s Center for Drug Evaluation and Research (CDER). Breakthrough therapy designation requests received by the CDER continue to increase over time, whereas the CBER numbers’ rise is slower and steadier, as shown in the tables below.
Table 1: CDER Breakthrough Therapy Designation Requests Received by Fiscal Year (Data as of September 30, 2018; Cohort: July 9, 2012)
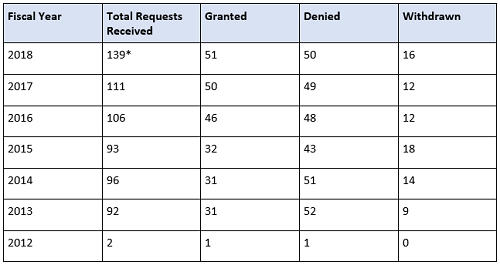
* Requests that are still pending a decision are included in the Total Requests Received column.
Table 2: CBER Breakthrough Therapy Designation Requests Received by Fiscal Year (Data as of September 30, 2018; Cohort: July 9, 2012)

* Requests that are still pending a decision are included in the Total Requests Received column. Withdrawn applications are counted in received total (for CBER).
The CBER House Rules: Recent Approvals
Although the breakthrough therapy designation track may appear to be faster, there are considerations an early-phase program must make. A breakthrough therapy designation conveys all of the fast-track program features, more intensive FDA guidance on an efficient drug development program, an organizational commitment involving senior managers, and eligibility for rolling review and priority review.
Several notable recent “combination therapy” approvals from the CBER include drugs for patients at the end of current beneficial doses. On February 26, 2018, the FDA approved abemaciclib (Verzenio) in combination with an aromatase inhibitor as initial endocrine-based therapy for postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced or metastatic breast cancer. On May 3, 2018, Andexxa was approved. This is indicated for patients treated with rivaroxaban and apixaban, when reversal of anticoagulation is needed due to life-threatening or uncontrolled bleeding.
Medication-Assisted Treatments
Another use of the identifier combination, which is outside the combination product sector and a part of the government’s response to the opioid epidemic, is the medication-assisted treatment (MAT). MAT is defined as the use of medications in combination with counseling and behavioral therapeutics which is effective in the treatment of opioid use disorders and can help some people sustain recovery. MAT3 falls under the Substance Abuse and Mental Health Services Administration.
FDA Commissioner Scott Gottlieb stated on September 20, 2017, “Medication-assisted treatment (MAT) …is one of the major pillars of the federal response to the opioid epidemic in this country. In fact, patients receiving MAT cut their risk of death from all causes in half. …”
Three drugs have been approved by the FDA for the treatment of opioid dependence: buprenorphine, methadone, and naltrexone. All three of these treatments have been demonstrated to be safe and effective in combination with counseling and psychosocial support.
Positive news about triple-drug therapies in clinical trials for cystic fibrosis treatment has just been published.4 Now identified as a “triple-combination treatment,” a combination of two available CTFR (cystic fibrosis transmembrane conductance regulator) modulators, tezacaftor and invacaftor, with two different experimental modulators per trial, VX-659 and VX-445, has demonstrated that “either of the two triple-drug regimens could potentially benefit 90 percent of people with cystic fibrosis.”5
Conclusion
As technology advances and experimentation with existing approved entities broadens, providing safe and longer-lasting options for patients will become a primary goal for all future product development. The use of “combination” will continue, and the proper use of it will depend on regulatory professionals reminding development teams of the differences as data becomes available. Strategic planning begins when management positions a concept, and the pathways the resulting search may take will depend on your regulatory questioning and innovative directions.
References:
May 30, 2018: Expanded approval of Xeljanz (tofacitinib) to include adults with moderately to severely active ulcerative colitis. Julie Beitz, director of the Office of Drug Evaluation III in the CDER, said: “Today’s approval provides an alternative therapy for a debilitating disease with limited treatment options.”
March 6, 2018: Trogarzo (ibalizumab-uiyk), a new type of antiretroviral medication for adult patients living with HIV who have tried multiple HIV medications in the past (heavily treatment-experienced) and whose HIV infections cannot be successfully treated with other currently available therapies (multi-drug resistant HIV, or MDR HIV).
August 3, 2107: Vyxeos is the first approved treatment specifically for patients with certain types of high-risk AML. According to Richard Pazdur, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the CDER: “Vyxeos combines two commonly used chemotherapies into a single formulation that may help some patients live longer than if they were to receive the two therapies separately.”
- Frequently Asked Questions: Breakthrough Therapies, https://www.fda.gov/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/FDASIA/ucm341027.htm
- Information about Medication-Assisted Treatment (MAT), https://www.fda.gov/Drugs/DrugSafety/InformationbyDrugClass/ucm600092.htm
- Oct. 18, 2018, New England Journal of Medicine; Steven Rowe, M.D., M.S.P.H., director, Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham; Michael Boyle, M.D., senior vice president, therapeutics, Cystic Fibrosis Foundation, Bethesda, Md.
- Triple-Drug Therapy Might Be Cystic Fibrosis “Breakthrough,” HealthDay (10/18, Norton) reports the results (10/18) of two preliminary trials published in the New England Journal of Medicine finding that “either of two triple-drug regimens could potentially benefit 90 percent of people with cystic fibrosis.” https://consumer.healthday.com/respiratory-and-allergy-information-2/cystic-fibrosis-news-167/3-drug-therapy-might-be-cystic-fibrosis-breakthrough-738758.html
About The Author:
 Lynn C. Hansen, RAC, director of regulatory affairs, Pharmatech Associates, has worked in the development and regulatory management of product programs within the pharmaceutical and bioscience industries for 30 years. Her experience spans the drug development lifecycle from product development and research through to commercial launch and post-market monitoring for solid dose, parenteral, and combination products. She has held leadership positions in both Big Pharma and virtual start-up organizations, overseeing the regulatory submission and maintenance programs for multiple products. Hansen’s expertise includes CMC, clinical, and non-clinical modules and extends to both U.S. and global regulatory filings that utilize the eCTD format. She is an active member of the Regulatory Affairs Professional Society certification program and currently sits on the Regulatory Affairs Certification Board.
Lynn C. Hansen, RAC, director of regulatory affairs, Pharmatech Associates, has worked in the development and regulatory management of product programs within the pharmaceutical and bioscience industries for 30 years. Her experience spans the drug development lifecycle from product development and research through to commercial launch and post-market monitoring for solid dose, parenteral, and combination products. She has held leadership positions in both Big Pharma and virtual start-up organizations, overseeing the regulatory submission and maintenance programs for multiple products. Hansen’s expertise includes CMC, clinical, and non-clinical modules and extends to both U.S. and global regulatory filings that utilize the eCTD format. She is an active member of the Regulatory Affairs Professional Society certification program and currently sits on the Regulatory Affairs Certification Board.